


FDA Home Page | CDRH Home Page | Search | CDRH
A-Z Index | Contact
CDRH
![]()
|
|
FDA Home Page | CDRH Home Page | Search | CDRH
A-Z Index | Contact
CDRH |
Guidance for Industry and FDA - Medical Glove Guidance Manual |
|
This guidance document is being distributed for comment
purposes only.
Draft released for comment on July 30th, 1999
 |
U.S. Department of Health and Human Services Division of Small Manufacturers, International and
Consumer Assistance |
Comments and suggestions regarding this draft document should be submitted by October 28th, 1999 to Docket No. 99D-2335, Dockets Management Branch, Division of Management Systems and Policy, Office of Human Resources and Management Services, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD 20852.
World Wide Web/CDRH home page at http://www.fda.gov/cdrh/manual/glovmanl.pdf or for instructions on how to obtain this manual use CDRH Facts on Demand at 1-800-899-0381 or 301-827-0111, specify number 852 when prompted for the document shelf number.
This document contains guidance on the basic regulatory requirements set forth in FDA’s regulations that all manufacturers and importers must consider when they plan to market medical gloves. It is important to know these regulatory requirements, how to determine which ones are pertinent to your particular situation, and the proper sequence for fulfilling them. This document contains guidance on establishment registration, device listing, labeling requirements, classification, premarket notification [510(k)], medical device reporting, and good manufacturing practices of significance to manufacturers and importers of medical gloves. To the extent this guidance discusses regulatory requirements, these are requirements established by the Federal Food, Drug and Cosmetic Act or FDA’s implementing regulations in Part 800 of Title 21 of the Code of Federal Regulations. This guidance incorporates changes required by the Food and Drug Administration Modernization Act of 1997.
This document is intended to replace publication FDA 97-4257, "Guidance for Medical Gloves: A Workshop Manual" after final comments are received and incorporated.
The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the Agency.
_________________________
1This guidance
document represents the Agency's current thinking on medical gloves. It does not
create or confer any rights for or on any person and does not operate to bind
FDA or the public. An alternative approach may be used if such approach
satisfies the requirements of the applicable statute, regulations, or both.
1 INTRODUCTION
HEALTH CARE WORKERS TO USE PROTECTIVE BARRIERS|
REGULATORY AND QUALITY DEVELOPMENTS
Historical Activities
Recent ActivitiesIdentity statement
Hypoallergenic claim
Chemical sensitivity
Reclassification
Protein levels
Powder-free
Powdered gloves
Expiration dating
2 OVERVIEW OF STATUTORY AND REGULATORY REQUIREMENTS
REGISTRATIONWho Must List
Importer's Obligation
Forms and Instructions
Where to MailPREMARKET NOTIFICATION [510(k)]
What is a 510(k) Premarket Notification Submission
Who Must Submit a Premarket Notification
Overview of a 510(k) Submission
Truthful and Accurate Statement
Indications for Use Statement
How Do You Submit a Premarket Notification for Medical Gloves
Safe Medical Devices Act Summary or Statement
Where Do You Submit the 510(k)
FDA Requests Additional Information
Modifications
"Special" 510(k)
Transfer of Ownership of a 510(k) *THE NEW 510(k) PARADIGM
LABELING
QUALITY SYSTEM REGULATION
MEDICAL DEVICE REPORTING
APPENDIX A - SUBSTANTIALLY EQUIVALENT LETTER
APPENDIX B - ACKNOWLEDGEMENT LETTER
3 PRODUCT IDENTIFICATION
EXAMINATION GLOVES (PATIENT)Table 3.1 Powdered Patient Examination Glove Proposed Classification
Dental Examination GlovesSPECIALTY/ CHEMOTHERAPY GLOVES
SURGEON'S GLOVESSurgeon’s Gloves, Special
Table 3.2 Powdered Surgeon's Glove Proposed Classification
Microsurgery Gloves
Orthopedic Surgeon’s Gloves
Autopsy Surgeon’s GlovesGLOVE LINERS / UNDERGLOVES
SURGEON’S GLOVING CREAM
RADIOGRAPHIC PROTECTION GLOVES
EMBALMING GLOVES
FOOD HANDLING GLOVES
CLEANING AND OTHER NON-MEDICAL GLOVES
MANUFACTURER NAME IMPLIES MEDICAL DEVICE
LEAK DETECTORS
4 GLOVE LUBRICANTS
RELEASE AGENTS
POWDERED GLOVES
CONTENT AND FORMAT OF PMAS FOR ABSORBABLE DUSTING POWDER FOR SURGICAL GLOVES
ABSORBABLE DUSTING POWDER, USP
FIRMS WITH NDA or PMA for U.S.P. ABSORBABLE DUSTING POWDER
FEDERAL REGISTER, VOL. 36, NO. 101 - TUESDAY, MAY 25, 1971
5 BIOCOMPATIBILITY
INTRODUCTION
SKIN IRRITATION AND DERMAL SENSITIZATION STUDIESPrimary Skin Irritation Test (Animal Study)
Dermal Sensitization Study (Animal Study)
Hypoallergenicity
Testing for Skin Sensitization to ChemicalsCOLOR AND FLAVOR ADDITIVES
NON-PYROGENIC
LABORATORIES THAT PERFORM ASTM DRAIZE TEST
PRIMARY SKIN IRRITATION AND HUMAN DERMAL TOXICITY TEST LABS
REFERENCE TO ASTM TEST LAB DIRECTORY
Name and Place of Business
Statement of Identity
Net Quantity of Contents Statement
Country of Origin
Adequate Directions for Use
Powder and Protein Labeling
Expiration Date
Lot Number
Donning Powder or Lubricant Identification
Standards
Bar Coding
National Health Related Items Code"Powder-Free"
Protein Label Claims
Polymer-Coated Gloves
Chemical Sensitization
Color and Flavor Additives
Chemotherapy Label Claim
Non-Pyrogenic
Hypoallergenicity
Special Label Claims *
7 STATEMENT AND SUMMARY INFORMATION
INTRODUCTION
TRUTHFUL AND ACCURATE STATEMENT AND FORMAT
INDICATIONS FOR USE STATEMENT AND SAMPLE FORMAT
510(k) SUMMARY OR STATEMENT
Use of Summaries and Statements
Who Responds to Requests for 510(k) InformationREQUIREMENTS FOR A 510(k) SUMMARY
REQUIREMENTS FOR A 510(k) STATEMENT AND SAMPLE FORMAT
8 PATIENT EXAMINATION GLOVES
DEFINITION AND REQUIREMENTS
VOLUNTARY STANDARDS
BIOBURDEN AND MOISTUREPREMARKET NOTIFICATION SUBMISSION FORMAT
New 510(k) Paradigm
Sample Format for A Premarket Notification [510(K)] for Examination Gloves
Premarket Notification 510(k) Submission Applicant
Truthful and Accurate Statement
Indications for Use Statement
Glove Proprietary or Trade Name
Name and Location of ACTUAL Manufacturer
Labels, Labeling, and Advertising
Classification Information
Specifications
Quality Assurance Testing
Sterility
FORMER Release Powder or Chemical
Dusting or Donning Powder
Weight of Powder-free Residue
Protein Level of Natural Rubber Latex Gloves
Protein Control
Chemical Sensitivity Claim
Color or Flavor Additives
Biocompatibility
Expiration Date or Quality at Delivery
Other Claims Requiring Data
510(k) Summary/Statement Requirement: *
9 SURGEON’S GLOVES
DEFINITION AND REQUIREMENTS
VOLUNTARY STANDARDS
STERILITY, BIOBURDEN AND MOISTURE
PREMARKET NOTIFICATION [510(k)] SUBMISSION FORMAT *New 510(k) Paradigm
Sample Format for a Premarket Notification [510(K)] for Surgeon’s Gloves
Premarket Notification 510(k) Submission Applicant
Sample Premarket Notification Truthful and Accurate Statement
Indications for Use Statement
Glove Proprietary or Trade Name
Name and Location of ACTUAL Manufacturer
Labels, Labeling, and Advertising
Classification Information
Specifications
Quality Assurance Testing
Sterility
Former Release Powder or Chemical
Absorbable Dusting or Donning Powder
Weight of Powder-free Residue
Protein Level of Latex Gloves
Protein Control
Chemical Sensitivity Claim
Color or Flavor Additives
Glove Biocompatibility
Expiration Date or Quality at Delivery
Other Claims Requiring Data
510(k) Summary/Statement Requirement
10 QUALITY SYSTEM REGULATORY REQUIREMENTS AS APPLIED TO MEDICAL GLOVES
INTRODUCTIONDynamic System
FDA Quality System Regulation (diagram)
Quality System & QS Audits (diagram)
Quality System Documents *Design and Development Planning
Design Control System Outline
Design Input *Documenting Design Output
Acceptance Criteria
Design Output ApprovalDesign Verification and Validation
Labeling Verification
Design Transfer
Design Changes
Design History File
Sample Design Input Requirements Procedure *Written Procedures
Sample Device Master Record Glove Specification
Record Retention *COMPONENTS AND MANUFACTURING MATERIALS
Component Qualification
Specifications
Supplier Evaluation
Acceptance Procedures
Acceptance Criteria
Testing and Inspection
Acceptance and Rejection Records
Obsolete, Deteriorated, and Rejected Product
Product Storage
Receiving History Record
Receiving Latex Test Data
Component and Material Status DecalsContamination Control
Environmental Control
Monitoring
Personnel PracticesEQUIPMENT AND MANUFACTURING MATERIALS *
Maintenance, Inspection, and Adjustment
Manufacturing Materials
Control UseReceipt and Release
Area Separation and Inspection
Storage
Packaging
Procurement, Acceptance and Storage
Packaging Process
Sample Label for Dispenser Box
Shipping for ProcessingPRODUCTION AND PROCESS CONTROL
Specifications
Processing ControlsPinholes
Processing Chemicals
Water-Soluble Proteins
Bioburden ControlReworking
Retesting
Sample Lab Analysis of Latex Compounding
Example Machine Dep Line Parameters Per DayEthylene Oxide (EtO )
Radiation Sterilization
Contract SterilizationComplaint Handling System
Complaint Responsibility
Complaint Records
Investigation Records and Location
Medical Device Reporting *
Sample Complaint Processing ProcedureCORRECTIVE AND PREVENTIVE ACTION
CHECKLIST FOR DESIGN OF NATURAL RUBBER LATEX GLOVES AND PROCESSES
11 COMPLIANCE ACTIVITIES
FACTORY INSPECTIONSSample of a letter to pre-announce an FDA International Inspection Letter
Conduct During the Inspection
Close-out Meeting
After the InspectionManagement Letter
Warning Letter
Seizure
Administrative Detention
Restraining Orders and InjunctionsFDA REQUIREMENTS FOR MEDICAL GLOVES
COMPLIANCE ACTIVITIES FOR IMPORTED GLOVES
DETENTION *Administrative Detention
Detention Without Physical Examination
Criteria for Detention Without Physical Examination (DWPE) - General
Criteria for Release from Detention Without Physical Examination (DWPE) - General
Import Alerts for Medical GlovesFDA SAMPLING EFFORTS
ENFORCEMENT STRATEGYAppendix A - Selections From the FDA Regulatory Procedures Manual
Appendix B - Sec.335.700 Surgeons' Gloves and Patient Examination Gloves
Appendix C - 21 CFR, Title 21, Volume 8
12 VOLUNTARY STANDARDS
INTRODUCTION TO STANDARDS
USE OF GLOVE RELATED STANDARDSLatex Patient Examination Glove Standards
Synthetic Material Patient Examination Glove Standards
Latex Surgical Glove Standards *
HEALTH CARE
WORKERS TO USE PROTECTIVE BARRIERS
REGULATORY AND
QUALITY DEVELOPMENTS
Identity Statement
Hypoallergenic Claim
Chemical Sensitivity
Reclassification
Protein Levels
Powder-Free
Powdered Gloves
Expiration Dating
QUALITY
SYSTEM
VOLUNTARY
STANDARDS
HEALTH CARE WORKERS TO USE PROTECTIVE BARRIERS1
The United States (U.S.) Centers for Disease Control (CDC) published a report on August 21, 1987, that emphasized the need for all health care workers to routinely use appropriate barrier precautions when contact with blood or other body fluids of any patient is anticipated.
On December 6, 1991, the U.S. Occupational Safety and Health Administration (OSHA) enacted regulations requiring the use of work practice controls and protective clothing, including gloves, to minimize worker exposure to blood-borne pathogens.
Subsequently, importation of medical gloves rose dramatically from 1986, when less than 1 billion gloves were imported, to 1997 when that number increased to about 23 billion. It is anticipated that gloves will be used increasingly to help prevent the transmission of Hepatitis B Virus (HBV), Human Immunodeficiency Virus (HIV) and other blood-borne pathogens.
________________________
1Information
on the regulatory requirements for patient examination gloves, surgeon's gloves,
and some non-medical gloves is contained in this manual. To the extent this
guidance discusses regulatory requirements, these are requirements established
by the Federal Food, Drug and Cosmetic Act or FDA’s implementing regulations in
Part 800 of Title 21 of the Code of Federal Regulations. Increased knowledge of
regulatory obligations will result in increased compliance if manufacturers are
willing to earnestly apply that knowledge. Although not specifically intended
for other devices made of latex, such as dental dams, manufacturers should find
that the general guidance in this manual is also helpful in meeting quality
requirements for these devices.
The CDC report recommends that health care workers wear medical gloves when:
Because of the emphasis in the CDC recommendations upon gloves as a barrier to HIV, HBV and other blood-and-fluid borne infectious agents, and the need for greater assurance against transmission between patients and health care workers, the Food and Drug Administration (FDA) believes that gloves worn by health care workers must provide an effective barrier to the transmission of infectious agents. Obviously, this effective barrier can be provided by ensuring that medical gloves meet appropriate standards and prevailing guidelines.
REGULATORY AND QUALITY DEVELOPMENTS
FDA’s regulations require that regulates medical gloves, requires that medical gloves be correctly labeled and cleared for marketing through a premarket notification submission [510(k)] prior to being distributed in the U.S. FDA’s regulations at Title 21 Code of Federal Regulations Part 820 also require manufacturers to produce gloves according to the Quality System (QS) regulation (formerly Good Manufacturing Practices regulation) to assure that gloves are produced at an acceptable quality level, thus helping to assure their safety and effectiveness. The safety and effectiveness of medical gloves can be compromised by many kinds of defects. These defects can be controlled or eliminated through proper quality control procedures. The Agency has determined that glove defects, such as pinholes, which are not readily detectable by the users of gloves, can significantly compromise the effectiveness of the barrier and result in patients or health care workers being unnecessarily exposed to infectious agents. In order to increase the level of public health protection, FDA has taken several historical and recent actions as summarized below.
From 1987 to the present, FDA has worked with manufacturers, standards groups, laboratories and the healthcare community to improve the safety and performance of gloves. The FDA:
A new regulation titled, "Natural Rubber-Containing Medical Devices; User Labeling" (http://www.fda.gov/cdrh/dsma/fr93097.html) became effective September 30, 1998 (see 21 CFR 801.437). The requirements of this regulation include the following two items:
Identity statement. The labeling of natural rubber latex devices must contain the statement, "Caution: This product contains natural rubber latex which may cause allergic reactions." This statement is also required by the proposed glove regulation.
Hypoallergenic claim. The labeling of natural rubber latex devices may no longer use the term "hypoallergenic."
Both of these requirements apply to all devices composed of or containing, or having packaging or components composed of or containing, natural rubber that contacts humans.
Chemical sensitivity. FDA has developed draft guidance for evaluating the chemical sensitization potential of medical devices containing latex and recommended labeling for products with reduced levels of chemical sensitizers. For guidance, please refer to the document titled, "Draft Guidance on the Content and Format of Premarket Notification [510(k)] Submissions for Testing for Skin Sensitization to Chemicals in Latex Products," available on the Internet at: http://www.fda.gov/cdrh/ode/944.html.
Additionally, FDA has prepared a proposed rule regarding medical gloves that will be published in the Federal Register. The main features of this rule are summarized as follows:
Reclassification. FDA is proposing the reclassification of surgeon's and patient examination gloves to from Class I Class II because general controls are insufficient to assure their safety and effectiveness. The proposed Class II glove types are:
Protein levels. FDA, industry, and ASTM have developed a standard that uses the modified Lowry method for measuring water-soluble proteins on finished latex gloves. This standard, D-5712, was approved by ASTM in April of 1995. As of May 1, 1995, manufacturers started filing 510(k) submissions with FDA to reflect optional claims for protein levels on glove labeling based on measurements made according to D 5712. ASTM is working to improve this standard.
FDA is proposing in the noted regulation that all surgeon's gloves and patient examination gloves bear labeling that states the upper limit of water extractable protein per glove and the upper limit recommended by FDA which is no more than 1200 µg per glove.
Powder-free. ASTM, FDA, and industry have developed a standard method for measuring the residual or trace powder level on "powder-free" gloves. This ASTM standard D 6124 covers former-release powders, donning powders and manufacturing debris.
FDA guidance (this manual) recommends that powder-free surgeon's gloves and patient examination gloves contain no more than 2 mg trace powder per glove.
Powdered gloves. ASTM, FDA and industry are developing a standard for measuring the donning powder on a powdered glove.
FDA is proposing in the noted regulation that all surgeon's gloves and patient examination gloves bear labeling that states the powder per glove and state the upper limit recommended by FDA which is proposed to be no more than 120 mg per glove.
Expiration dating. FDA is proposing in the noted regulation that all surgeon's and patient examination gloves bear an expiration date that is supported by stability studies demonstrating acceptable physical and mechanical integrity during the shelf life.
FDA is emphasizing that to meet requirements in the QS regulation, manufacturers should implement controls to minimize:
In addition to meeting regulatory requirements, medical gloves should conform to national voluntary consensus standards developed by industry, FDA and the:
American Society For Testing and Materials (ASTM)
100 Barr Harbor Drive
West Conshohocken, Pennsylvania 19428 USA
Phone: 610-832-9500
FAX: 610-832-9555
The ASTM standards for each type of glove is noted in appropriate sections of this manual. ASTM and other glove standards are listed in chapter 12.
Who Must List
Importer's Obligation
Forms and Instructions
Where to Mail
PREMARKET NOTIFICATION [510(k)]
What is a 510(k) Premarket Notification Submission
Who Must Submit a Premarket Notification
Overview of 510(k) Submission
Truthful and Accurate Statement
Indications for Use Statement
How Do You Submit a Premarket Notification for Medical Gloves
Safe Medical Devices Act Summary or Statement
Where Do You Submit the 510(k)
FDA Requests Additional Information
Modifications
"Special 510(k)"
Transfer of Ownership of a 510(k)
THE NEW 510(k)
PARADIGM
LABELING
QUALITY SYSTEM
REGULATION
MEDICAL DEVICE
REPORTING
APPENDIX A -
SUBSTANTIALLY EQUIVALENT LETTER
APPENDIX B -
ACKNOWLEDGEMENT LETTER
Medical devices are classified by the FDA into one of three regulatory Classes: I, II, or III as required by the FD&C Act. The class of a device determines the level of regulatory control that applies to it. Medical gloves are in Class I (currently, Class I reserved). In addition, FDA has proposed that surgeon's gloves and patient examination gloves be reclassified into Class II.
Medical gloves are subject to general controls as follows:
Regulations discussing these general controls are published in Title 21 of the United States (U.S.) Code of Federal Regulations (CFR). Applicable parts of these regulations and guidance on how to meet them are described herein.
REGISTRATION (21 CFR Part 807)
Who Must Register. All domestic and foreign medical glove manufacturers, contract manufacturers of finished gloves, specifications developers, contract sterilizers, initial domestic distributors (importers), repackers, and relabelers are required to register their establishment with FDA. To register, complete form FDA 2891, Initial Registration of Medical Device Establishment. You must use an original form -- do not use a photocopy. Forms can be obtained at FDA offices throughout the U.S. (please check your local telephone directory under Government) or by contacting the Division of Small Manufacturers Assistance (DSMICA) by FAX at 301-443-8818 or Email at dsma@cdrh.fda.gov. Be sure to include a clearly printed return mailing address.
When domestic and foreign manufacturers register for the first time, they must also submit a device listing form. Requirements, such as listing, 510(k) submissions and medical device reporting are described below. If you have filed or plan to file a 510(k), you do not need to submit the registration and listing forms until after you receive your marketing clearance letter from FDA.
Where to Mail. Mail the completed registration form FDA 2891 to the following address:
Information Processing and Office Automation Branch (HFZ-307)
Center for Devices and Radiological Health
Food and Drug Administration
2098 Gaither Road
Rockville, Maryland 20850 USA
FAX 301-495-4660 (For registration problems only -- do not FAX the form.)
Who Must List. Domestic manufacturers, foreign manufacturers, repackers, or relabelers must list with FDA the type of device they market in the U.S. Also, specifications developers are required to list each type of medical glove if they distribute medical gloves. To list, complete form FDA 2892, "Device Listing," and mail it to the above address. Use an original form -- do not use a photocopy and do not FAX the form to the FDA. (Information needed for Block #8, Classification Number, on form FDA 2892 can be located in Tables 3.1 and 3.2 under "Product Codes" in Chapter 3 of this guidance.)
Importer's Obligation. Initial distributors/importers have a listing obligation, but they do not satisfy it by completing form FDA 2892. Instead, initial distributors/importers should send a letter on their company letterhead to the above address.
The initial distributor’s listing letter must state:
Forms and Instructions. Blank copies of the establishment registration form FDA 2891, and the medical device listing form FDA 2892, along with the instruction booklet, are available from the Information Processing and Office Automation Branch at the above address or from:
Publications
DSMICA (HFZ-220)
Food and Drug Administration
1350 Piccard Drive
Rockville, Maryland 20850 USA
Please request by FAX at 301-443-8818. (Please make sure your FAX number, name and address are in large clear print on your FAX requests so the forms can be mailed to you.)
Where to Mail. Do not mail completed registration or listing forms to DSMICA. This will delay processing of the forms that you submit to FDA. Completed forms FDA 2891 and FDA 2892 should be mailed to the Information Processing and Office Automation Branch. After making a copy for your files, submit all pages of the original registration form and listing form to the Information Processing and Office Automation Branch, the same office and address shown under "Registration" on a previous page. Do not send photocopies or FAX copies. Only the original forms will be accepted for processing.
PREMARKET NOTIFICATION [510(k)] (21 CFR Part 807)
The following information is for your use in preparing a premarket notification [510(k)] for medical gloves. Although some of the information below may not be captured in the regulations, the suggestions represent FDA's position in rendering a substantial equivalence decision for a 510(k) for medical gloves.
What is a 510(k) Premarket Notification Submission. A 510(k) is a premarket application sent to the FDA documenting that the finished medical glove you wish to market is as safe and effective as a legally marketed medical glove that was, or is, on the U.S. market.
A premarket notification submission, also known as a 510(k) submission, must be submitted to FDA prior to marketing medical gloves as required by Section 510(k) of the FD&C Act. Upon receipt, FDA will send the manufacturer an acknowledgment letter which contains a unique document control number (i.e., K followed by 6 digits) that has been assigned to their 510(k) application. The acknowledgment letter with the 510(k) document control number (Attachment A in this chapter) is not clearance from FDA for the manufacturer to market the gloves. The manufacturer should not market or enter the gloves into the U.S. until a marketing clearance letter, also called an order or substantial equivalence letter, is received from FDA (Attachment B).
The premarket notification submitted to FDA must contain information that demonstrates that the glove is substantially equivalent to a medical glove that has been legally marketed in the U.S. which did not require a Premarket Approval Application (PMA). The submission should be adequate if it contains the data and information covered by the suggested 510(k) format outlined in Chapters 8 or 9. If the gloves conforms to a standard which has been recognized by the FDA, the standard becomes the basis for comparison.
Test data in 510(k) submissions should be the result of tests performed on finished medical gloves that were made by the same process as regular production medical gloves intended to be distributed. The water leak test data in the 510(k) submission should be from recently manufactured gloves and from gloves that have been aged for 3 to 12 months of real time aging or subjected to accelerated aging for 7 days at 70 degrees Centigrade as described in ASTM D standard 3578, D 3577 or D 5250, as appropriate, or an equivalent aging method. (In contrast, water leak testing for routine production is done on non-aged gloves.)
Who Must Submit a Premarket Notification. The following owners or operators must submit a 510(k) to the FDA:
After being cleared for commercial distribution by the FDA, the specific glove covered by that 510(k) may be imported by any one or more U.S. distributor(s). Only one premarket notification [510(K)] is required for each glove type (such as powder free or powdered, colored, flavored, protein content, claim, etc.). It is the responsibility of the distributor to provide the correct 510(k) number to the FDA upon request by the Agency.
Overview of a 510(k) Submission. It is extremely important that you follow the guidance in this manual and 21 CFR Part 807 when preparing and submitting a 510(k) for medical gloves because the submission must meet requirements for applicant and device identification, safety, performance, labeling, identification of intended use, public release of non-confidential information under Freedom of Information, etc. Details are found in Chapters 2 through 9 of this manual.
Truthful and Accurate Statement. As required by 21 CFR 807.87(j), all 510(k) submitters must include a statement that all data submitted must be truthful and accurate. The following language in the statement cannot be altered or modified.
I certify that, in my capacity as ( The Position Held in Company ) of ( Manufacturer’s Name ), I believe to the best of my knowledge, that all data and information submitted in the premarket notification are truthful and accurate and that no material fact has been omitted.
The statement should be signed by a responsible person of the firm required to submit the premarket notification -- not by a consultant for the firm submitting the premarket notification (see chapter 7 for details.)
Indications for Use Statement. The 510(k) submission must include an "Indications for Use" page that contains the name of the device and the indications for use of the gloves as described in Chapter 7 of this manual.
The information, data and labeling claims in the entire 510(k) submission should support and agree with the Indications for Use statement.
How Do You Submit a Premarket Notification for Medical Gloves. There is no form for submitting a 510(k) for medical devices, only a detailed format to follow as listed in 21 CFR Part 807. However, due to the large number of 510(k) submissions for medical gloves, the use of a uniform format specifically intended for medical glove submissions will reduce submission errors and make processing by FDA more efficient. A copy of a recommended "format" is contained in Chapter 8, Patient Examination Gloves. Similarly, a recommended format detailing the content of a 510(k) submission for surgeon’s gloves is included in Chapter 9, Surgeon’s Gloves.
Safe Medical Devices Act Summary or Statement. Persons who submit a 510(k) submission are required by the FD&C Act, as amended by the Safe Medical Devices Act of 1990 (SMDA 90), to provide to FDA as part of the 510(k) submission:
The requirements for the summary or statement are specific and detailed. See chapter 7 and 21 CFR §§807.92 and 807.93 for details and model language.
Where Do You Submit the 510(k). The original premarket notification submission and one copy should be sent by a method that assures a return receipt as proof of delivery. Send your 510(k) to the following address:
Document Mail Center (HFZ-401)
Center for Devices and Radiological Health
Food and Drug Administration
1390 Piccard Drive
Rockville, Maryland 20850 USA
It is illegal to place a device into commercial distribution in the U.S. until you receive a letter from FDA stating that your device is substantially equivalent. Marketing the device prior to FDA clearance would render the device adulterated under §501(f)(1)(B) of the FD&C Act and subject to enforcement action by FDA.
FDA Requests Additional Information. After you submit your application, if FDA requests additional information by telephone, FAX, Email, or letter, you should:
Modifications. Under the New 510(k) Paradigm, a manufacturer should refer to 21 CFR 807.81(a)(3) and the FDA guidance document entitled, "Deciding When to Submit a 510(k) for a Change to an Existing Device" at http://www.fda.gov/cdrh/ode/510kmod.html to decide if a device modification may be implemented without submission of a new 510(k). If a new 510(k) is needed for the modification and if the modification does not affect the intended use of the device or alter the fundamental scientific technology of the device, then summary information that results from the design control process can serve as the basis for clearing the application.
"Special" 510(k). The Center for Devices and Radiological Health has developed an alternative method for obtaining clearance to market a modified version of an existing, legally marketed, device. This method of submission is called the Special 510(k), as discussed in the next paragraph. A properly prepared Special 510(k), which is accepted by FDA, will be reviewed within 30 days. If, for some reason, FDA does not accept the submission as a Special 510(k) (e.g., new indication for use, your certifications are not done, etc.), it will be converted to a Traditional 510(k)
A manufacturer who is intending to modify his/her own legally marketed device will conduct the risk analysis and the necessary verification and validation activities to demonstrate that the design outputs of the modifed device meet the design input requirements. Once the manufacturer has ensured the satisfactory completion of this process, a Special 510(k): Device Modification may be submitted. The Special 510(k) is explained in two guidance documents, "The New 510(k) Paradigm - Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications," at: http://www.fda.gov/cdrh/ode/parad510.html and its companion document which shows examples, "Frequently Asked Questions On The New 510(k) Paradigm," also on FDA's web site at: http://www.fda.gov/cdrh/ode/qanda510k.pdf.
A 510(k) application for a modification must be complete. Do not state that the necessary information is in another 510(k); instead, include all the necessary information in your submission. Also, the 510(k) should include appropriate supporting data to show that the manufacturer has considered what consequences and effects the change or modification or new use might have on the safety and effectiveness of the gloves, as described in 21 CFR 807.87(g).
The description of the modified gloves should include differences from the predicate gloves that could significantly affect safety and effectiveness. Provide any animal, engineering, design verification, bench, clinical, functional, in vitro, chemical resistance, and/or any other testing data that support the claims in your labeling for your modified gloves.
The requirements described above for a modification would be fulfilled if the applicant supplies the new information in another complete submission using the format described in Chapter 8, Patient Examination Gloves. Similarly, a suggested format detailing the content of a 510(k) submission for surgeon's gloves is included in Chapter 9, Surgeon's Gloves. Also, the applicant should reference the 510(k) number for the original gloves or accessory.
Transfer of Ownership of a 510(k)
A premarket notification [510(k)], like any other piece of property, may be bought, sold, or otherwise transferred. After a 510(k) substantial equivalence determination is issued to the submitter by FDA, the FDA is not involved in subsequent transfer of ownership or questions of ownership of a 510(k). Consequently, the change of ownership is not submitted to the FDA.
Information documenting the transfer of ownership of a 510(k), including any legal transactions that transpired, should be maintained in the new owner's 510(k) files. Upon inspection of the firm or upon entry of glove shipments into the U.S., FDA may request a review of this documentation, and if the owner fails to provide such information, FDA may request the owner to submit a 510(k). Under these conditions the owner may not distribute the device until FDA clears the new submission.
The new owner of the transferred 510(k) should submit a new medical device listing, form FDA 2892, to FDA. The previous owner of the 510(k) should send:
1) a letter notifying FDA if they are now out of business, and
2) device listing forms deleting any listings for products no longer being marketed by that firm.
Please note that neither establishment registration nor medical device listing identifies the establishment with the 510(k) ownership. It is the responsibility of the new owner of the 510(k) to keep documentation proving ownership of the 510(k) in their files.
In order to avoid problems upon import of a device for which 510(k) ownership has been transferred, it is recommended that a copy of the key information about the ownership sale or transfer documentation accompany all shipments to the United States. It could be a simple, one-page document giving concise information detailing the transfer transaction.
A 510(k) submission for a new glove or for a modification to an existing glove may be submitted according to the guidance titled, "The New 510(k) Paradigm - Alternative Approaches to Demonstrating Substantial Equivalence in Premarket Notifications," available on the World Wide Web at: http://www.fda.gov/cdrh/ode/parad510.pdf
The FD&C Act defines a label as a "display of written, printed, or graphic matter upon the immediate container of any article" (i.e., glove dispenser box) (21 USC 321 (k)).
Labeling is a broader term defined by the FD&C Act as "all labels and other written, printed, or graphic matter" upon any article or any of its containers or wrappers; or accompanying the article at any time while it is being held for sale after shipment or delivery for shipment in interstate commerce [21 USC 321 (m)]. Labeling includes some advertising, brochures and instructions, in any media such as printed text, software, encoded disks, or electronic transmissions.
Title 21 CFR Part 801 details labeling requirements for medical devices. Labeling requirements for latex medical gloves is currently found in §801.437. Proposed labeling requirements for medical gloves are located in proposed §801.440 and in Chapter 6 of this guidance. Any 510(k) submissions for medical gloves which do not include samples of the labeling will not be reviewed by FDA. Labeling does not need to be in final printed format; draft labeling may be submitted. The final labeling, however, should be consistent with draft labeling submitted in the 510(k) and should agree with your drawings for labeling and preprinted packaging in your Quality System device master record.
Manufacturers of medical gloves are required to meet the current Quality System regulation for medical devices (21 CFR Part 820). The Quality System regulation requires that every finished medical glove manufacturer shall prepare and implement a quality assurance (QA) program or quality system that is appropriate to the specific type of glove being manufactured, and that meets the requirements of the QS regulation. A manufacturer's quality system must include:
Note that these quality system requirements are much more extensive than pass or fail inspections and air testing of finished gloves. They are intended to assure that continuing quality is incorporated into the gloves during manufacture, rather than by testing and removing defective gloves to achieve a quality product after manufacture. These requirements are discussed in more detail in Chapter 10, Quality System Requirements as Applied to Medical Gloves, with emphasis on latex processing.
The purpose of the Medical Device Reporting (MDR) regulation, found in 21 CFR Part 803, is to provide the FDA with postmarketing information regarding adverse events occurring with the use of medical devices. Under the current provisions of the MDR regulation, device user facilities, domestic distributors, importers, and both domestic and foreign manufacturers of medical devices are subject to certain MDR requirements.
Manufacturers, importers, and user facilities must report adverse events when a device has or may have caused or contributed to a death or serious injury, and must establish and maintain adverse event files. They must submit to FDA specified follow-up and summary reports. Manufacturers and importers are also required to report certain device malfunctions to the FDA. Domestic distributors of medical devices in the U.S. are only required to maintain incident files and no longer have a reporting requirement.
Information gathered through medical device reporting assists FDA in protecting the public health by helping to assure that devices are not adulterated or misbranded, and are safe and effective for their intended use. FDA uses the MDR information to determine if user education programs are needed, whether product labeling needs improvement, whether devices need to be recalled, and during premarket submission reviews.
Manufacturers and user facilities will find a variety of guidance documents and other useful information on the CDRH home page at: http://www.fda.gov/cdrh/index.html. For additional information on MDR requirements, visit the Medical Device Reporting home page at: http://www.fda.gov/cdrh/mdr.html. Instructions for Completing the Medical Device Reporting Annual User Facility Report, Form FDA 3419 are at: http://www.fda.gov/cdrh/3419inst.html.
APPENDIX A - SUBSTANTIALLY EQUIVALENT LETTER

APPENDIX B - ACKNOWLEDGEMENT LETTER
| DEPARTMENT OF HEALTH AND HUMAN SERVICES | Public Health Service Food and Drug Administration Center for Devices and Radiological Health |
Office of Device Evaluation
Document Mail Center (HFZ-401)
9200
Corporate Blvd.
Rockville, Maryland 20850
| [ Company Name ] | 510(k)
Number:___________________ Received:________________________ Product: _________________________ |
The Center for Devices and Radiological Health (CDRH), Office of Device Evaluation (ODE), has received the Premarket Notification you submitted in accordance with Section 510(k) of the Federal Food, Drug, and Cosmetic Act (Act) for the above referenced product. We have assigned your submission a unique 510(k) number that is cited above. Please refer prominently to this 510(k) number in any future correspondence that relates to this submission. We will notify you when the processing of your premarket notification has been completed or if any additional information is required. YOU MAY NOT PLACE THIS DEVICE INTO COMMERCIAL DISTRIBUTION UNTIL YOU RECEIVE A LETTER FROM FDA ALLOWING YOU TO DO SO.
On January 1, 1996, FDA began requiring that all 510(k) submitters provide on a separate page and clearly marked "Indication For Use" the indication for use of their device. If you have not included this information on a separate page in your submission, please complete the attached and amend your 510(k) as soon as possible. Also if you have not included your 510(k) Summary or 510(k) Statement, or your Truthful and Accurate Statement, please do so as soon as possible. There may be other regulations or requirements affecting your device such as Postmarket Surveillance (Section 522(a)(1) of the Act) and the Device Tracking regulation (21 CFR Part 821). Please contact the Division of Small Manufacturers Assistance (DSMICA) at the telephone or web site below for more information.
Please remember that all correspondence concerning your submission MUST be sent to the Document Mail Center (HFZ-401) at the above letterhead address. Correspondence sent to any address other than the Document Mail Center will not be considered as part of your official premarket notification submission. Because of equipment and personnel limitations, we cannot accept telefaxed material as part of your official premarket notification submission, unless specifically requested of you by an FDA official. Any telefaxed material must be followed by a hard copy to the Document Mail Center (HFZ-401).
You should be familiar with the manual entitled, "Premarket Notification 510(k) Regulatory Requirements for Medical Devices" available from DSMICA. If you have other procedural or policy questions, or want information on how to check on the status of your submission (after 90 days from the receipt date), please contact DSMICA at (301) 443-6597 or its toll-free number (800) 638-2041, or at their Internet address http://www.fda.gov/cdrh/dsma/dsmamain.html or me at (301) 594-1190.
| Sincerely yours, |
| Marjorie Shulman Consumer Safety Officer Premarket Notification Staff Office of Device Evaluation Center for Devices and Radiological Health |
Powdered Patient Examination Glove Classification (Table 3.1)
Dental Examination Gloves
SPECIALTY/
CHEMOTHERAPY GLOVES
SURGEON’S
GLOVES
Surgeon’s Gloves, Special
Powdered Surgeon’s Glove Classification (Table 3.2)
Microsurgery Gloves
Orthopedic Surgeon’s Gloves
Autopsy Surgeon’s Gloves
GLOVE LINERS /
UNDERGLOVES
SURGEON’S GLOVING
CREAM
RADIOGRAPHIC
PROTECTION GLOVES
EMBALMING
GLOVES
FOOD
HANDLING GLOVES
CLEANING AND OTHER
NON-MEDICAL GLOVES
MANUFACTURER NAME
IMPLIES MEDICAL DEVICE
LEAK
DETECTORS
Classified medical gloves, accessories to gloves, and a few industrial gloves are briefly described in this chapter. The classification names and numbers for these medical devices are listed in Tables 3.1 and 3.2 because this information is needed when assembling a 510(k) submission. All references listed below are to Title 21 of the Code of Federal Regulations (CFR).
Under the proposed l999 rule, patient examination gloves would be classified as follows:
§880.6250 Patient examination gloves, powdered.
(a) Identification. A powdered patient examination glove is a disposable device made of natural rubber latex or synthetic material that bears powder to facilitate donning and is intended to be worn on the hand or finger(s) for medical purposes to provide a barrier against potentially infectious materials and other contaminants.
(b) Classification. Class II special controls are as follows:
(1) Guidance document. The Center for Devices and Radiological Health, FDA, "Medical Glove Guidance Manual,'' revised July 1999.
(2) Labeling. User labeling requirements in §801.440 of this chapter.
§880.6251 Patient examination gloves, powder-free.
(a) Identification. A powder-free patient examination glove is a disposable device made of natural rubber latex or synthetic material that may bear a trace amount of glove powder and is intended to be worn on the hand or finger(s) for medical purposes to provide a barrier against potentially infectious materials and other contaminants.
(b) Classification. Class II special controls are as follows:
(1) Guidance document. The Center for Devices and Radiological Health, FDA, ``Medical Glove Guidance Manual,'' revised July 1999.
(2) Labeling. User labeling requirements in §801.440 of this chapter.
Examination gloves are proposed for reclassification into Class II, and if they become class II, new or modified examination gloves will be required to meet the design controls in §820.30 of the QS regulation.
Table 3.1 POWDERED Patient Examination Glove Proposed Classification
|
Common Name |
Product Code |
*21 CFR
Classification |
CLASS** |
| Vinyl (PVC) | 80LYZ | 880.6250 | II |
| Latex | 80LYY | 880.6250 | II |
| Polymer (Nitrile, Polyurethane, etc.) | 80LZA | 880.6250 | II |
| Finger Cot | 80LZB | 880.6250 | II |
| Specialty/ Chemotherapy Gloves | 80LZC | 880.6250 | II |
POWDER-FREE Patient Examination Glove Proposed Classification
|
Common Name |
Product Code |
*21 CFR
Classification |
CLASS** |
| Vinyl (PVC) | 80LYZ | 880.6251 | II |
| Latex | 80LYY | 880.6251 | II |
| Polymer (Nitrile, Polyurethane, etc.) | 80LZA | 880.6251 | II |
| Finger Cot | 80LZB | 880.6251 | II |
| Specialty/ Chemotherapy | 80LZC | 880.6251 | II |
(Dental, special, and chemotherapy are adjectives modifying examination -- Thus, any glove in the above list could be manufactured and labeled as a dental, special, or chemotherapy examination glove to meet the needs of users.) * The information in this table is for gloves that meet the description in their classification regulation and, in the case of examination gloves, meet American Society for Testing and Material (ASTM) standard D-3578, D-5250 or an equivalent standard. The ASTM standard for finger cots is D-3772. (The ASTM standard for nitrile gloves should be published in 1999.) ** Until the effective date of a final rule reclassifying patient examinations gloves, they remain in Class I. | |||
Dental Examination Gloves. Gloves worn during dental cleaning, filling and the like are patient examination gloves and such gloves must meet the requirements for patient examination gloves. The term "dental" may be used in the labeling of gloves intended for dentistry. See the classification information in Table 3.1 under examination gloves. Dental examination gloves are usually "powder-free."
Gloves used for dental surgery are surgeon’s gloves and must meet the requirements for surgeon’s gloves. The term "dental" may be used in the labeling of gloves intended for dental surgery. Gloves for dental surgery may be thicker than standard surgeon’s gloves. The labeling may contain the thickness of the gloves but ambiguous terms such as "extra thick" are not acceptable to the FDA.
SPECIALTY/ CHEMOTHERAPY GLOVES
Chemotherapy gloves are specialty medical examination gloves and require premarket notification [510(k)] clearance from FDA before marketing. Chemotherapy gloves should meet the ASTM standard D 3578 or an equivalent standard for examination gloves; however, they are usually 0.10 mm or more in thickness which is more than the 0.08 mm minimum allowed for examination gloves.
In l999 FDA proposed in a rule in the Federal Register notice to reclassify surgeon's gloves into Class II Surgeon's Glove, Powdered, and Surgeon's Glove, Powder-free, because general controls are insufficient to assure their safety and effectiveness. Class II allows the use of special controls. The proposed classification regulations are as follows:
§878.4460 Surgeon's gloves, powdered.
(a) Identification. A powdered surgeon's glove is a disposable device made of natural rubber latex or synthetic material that bears powder to facilitate donning, and it is intended to be worn on the hands, usually in surgical settings, to provide a barrier against potentially infectious materials and other contaminants. The lubricating or dusting powder used on these gloves is classified separately in §878.4480.
(b) Classification. Class II special controls are as follows:
(1) Guidance document. The Center for Devices and Radiological Health, FDA, "Medical Glove Guidance Manual,'' revised July 1999.
(2) Labeling. User labeling requirements in §801.440 of this chapter (i.e., 21 CFR §801.440).
§878.4461 Surgeon's gloves, powder-free.
(a) Identification. A powder-free surgeon's glove is a disposable device made of natural rubber latex or synthetic material that may bear a trace amount of glove powder and is intended to be worn on the hands, usually in surgical settings, to provide a barrier against potentially infectious materials and other contaminants.
(b) Classification. Class II special controls are as follows:
(1) Guidance document. The Center for Devices and Radiological Health, FDA, "Medical Glove Guidance Manual,'' revised July 1999.
(2) Labeling. User labeling requirements in §801.440 of this chapter (i.e., 21 CFR §801.440).
Biocompatibility data for finished sterile gloves should be submitted in a 510(k) submission and also filed in the Quality System design history file per 21 CFR §820.30 to demonstrate that the gloves are safe for the intended use. Various manufacturing materials are added to latex and polymer mixtures to aid in processing, improve glove performance, improve glove stability, etc. Some of these may be adverse materials that have the potential to cause irritation, impair wound healing or other problems. Adverse manufacturing material residues that affect compromised tissue, mucous membranes or skin must be removed or limited as required by §820.3(p) and §820.70(h) of the Quality System regulation and by your labeling claims.
Surgeon’s gloves must be distributed sterile. FDA will not accept a 510(k) for a non-sterile surgeon’s glove. The shipment of medical gloves to and from a contract sterilizer is regulated under the labeling requirements in 21 CFR §801.150(e). (See Chapter 10.)
Surgeon’s Gloves, Special. Surgeon’s gloves with attributes for special applications with attached or integrated accessories must meet the basic regulatory requirements for surgeon’s gloves as outlined above. In addition, any accessory must meet the manufacturer’s labeling claims, be safe and effective (have clinical utility) and meet all other regulatory requirements. If the glove and accessory is substantially equivalent to a glove and an accessory that is already cleared for commercial distribution by the 510(k) process, then a 510(k) for the combination should be submitted to ODE. Otherwise, a Premarket Approval (PMA) may be required. Please consult with the Division of Small Manufacturers, International and Consumer Assistance (DSMICA), phone 800-638-2041, before preparing a PMA.
Table 3.2 POWDERED Surgeon's Glove Proposed Classification
| Common Name | Product Code |
*21 CFR Classification Number |
Class*** |
| Surgeon’s Gloves |
79KGO |
878.4460 |
II |
| Surgeon’s Glove with/or accessory** |
79KGO |
878.4460 |
II** |
| Microsurgery Gloves |
79KGO |
878.4460 |
II |
| Orthopedic Surgeon’s Gloves |
79KGO |
878.4460 |
II |
| Autopsy Surgeon’s Gloves |
79KGQ |
878.4460 |
II |
| Surgeon’s Gloving Cream |
9KGQ |
878.4470 |
I Exempt |
| Glove Liners/Undergloves |
79KGO |
878.4460 |
II |
| Leak Detectors & Glove Accessories |
79LDQ |
878.4460 |
II |
POWDER-FREE Surgeon's Glove Proposed Classification
| Common Name | Product Code |
*21 CFR Classification Number |
Class*** |
| Surgeon’s Gloves |
79KGO |
878.4461 |
II |
| Surgeon’s Glove with/or accessory** |
79KGO |
878.4461 |
II** |
| Microsurgery Gloves |
79KGO |
878.4461 |
II |
| Orthopedic Surgeon’s Gloves |
79KGO |
878.4461 |
II |
| Autopsy Surgeon’s Gloves |
79KGQ |
878.4461 |
II |
| Glove Liners/Undergloves |
79KGO |
878.4461 |
II |
| Leak Detectors & Glove Accessories |
79LDQ |
878.4461 |
II |
* The information in Table 3.2 is for gloves that meet the description in the classification regulation and, in the case of surgeon’s gloves, meet ASTM standard D 3577 or an equivalent standard. ** The accessory may have a different classification -- contact DSMICA by FAX at 301-443-8818 or contact ODE by phone at 301-443-8879 for case-by-case guidance. *** Until the effective date of a final rule reclassifying surgeon's, they remain in Class I. | |||
Microsurgery Gloves. Microsurgery gloves are surgeon’s gloves that meet the ASTM standard D 3577 for thickness and other parameters but are carefully processed so as to have a thickness, particularly at the fingertips, that is near the minimum allowed by ASTM D 3577. A 510(k) submission should contain all of the information applicable to regular surgeon’s gloves. FDA does not accept 510(k) submissions for microsurgery gloves that are thinner than allowed by the ASTM standard.
Orthopedic Surgeon’s Gloves. Orthopedic surgeon's gloves are a special form of surgeon’s gloves and must meet the requirements for surgeon’s gloves. Orthopedic surgical gloves may be thicker and more resistant to tear than other surgical gloves. The thickness and other parameters of orthopedic gloves may be stated in the labeling; whereas terms such as "extra thick," "super strong," etc., are ambiguous and the use of such terms results in a device being misbranded. The minimum biocompatibility tests are skin irritation and dermal sensitization.
Autopsy Surgeon’s Gloves. Autopsy gloves are a special form of surgeon’s gloves intended for use during autopsy procedures and require premarket notification [510(k)] clearance from FDA before marketing. Some autopsy surgeon’s gloves may be similar to orthopedic surgeon’s gloves. Pinhole, labeling, donning powder or lubricant, protein, manufacturing material residues, and powder-free requirements for autopsy gloves are the same as for surgical gloves. The minimum biocompatibility tests are skin irritation and dermal sensitization.
Glove liners or undergloves are worn with patient examination or surgeon’s gloves, and may be made of materials such as cotton to prevent the medical glove from contacting the user’s hand or may be made of materials that are resistant to cutting or puncture. Added protection is provided by reducing the risk of a cut or puncture wound during surgical or examination procedures, absorbing perspiration, and by reducing the potential for skin irritation. Glove liners and undergloves are accessories to medical gloves and are classified the same as the gloves. Currently, they are Class I devices.
Because glove liners and undergloves contact the skin, biocompatibility data should be submitted with a 510(k) to show that they are safe for the intended use (See Chapter 5, Biocompatibility). When glove liners are made of clean, non-coated, common textiles, biocompatibility data is not needed. Manufacturers of accessories such as glove liners are required to submit a premarket notification [510(k)], register their establishment, list the glove liners, meet the medical device Quality System regulation, and properly label their glove liners. If a manufacturer claims their glove liners are leakproof, then the glove liners have to meet the ASTM acceptable quality limit (AQL) for pinholes.
Surgeon’s gloving cream is intended to lubricate the user’s hand before putting on a surgeon’s glove. This cream may also be used with examination gloves.
Gloving cream is classified under 21 CFR 878.4470 as a Class I device. Gloving cream was exempted from premarket notification requirements by a notice in the Federal Register, Vol. 59, page 63010, December 7, 1994. If the intended use of the cream is different from that described in 21 CFR 878.4470, i.e., "…lubricating the user's hand…," the cream is not exempt from the 510(k) requirements.
Gloving creams should be safe and effective and should not degrade the glove material in latex or other gloves, i.e., the creams should not be oil-based. If manufacturers modify the ingredients of an existing gloving cream or introduce a new gloving cream into commercial distribution, such manufacturers are cautioned that the cream should perform as claimed, and they should have biocompatibility data on file to show that the new or modified cream is safe for the intended use. (Medical devices including gloving cream in commercial distribution in the U.S. are never exempt from the adulteration and misbranding provisions and penalties of the FD&C Act.)
RADIOGRAPHIC PROTECTION GLOVES
Radiographic protection gloves are classified as Class I devices currently exempt from premarket notification under 21 CFR 892.6500 as "personnel protective shield." These devices are intended to protect the operator, patient or other person from unnecessary exposure to radiation during radiological procedures by providing an attenuating barrier to radiation. The generic type of device includes articles of clothing such as gloves.
These gloves should meet the FDA or an equivalent water leak test and the minimum biocompatibility tests such as skin irritation and dermal sensitization. Manufacturers of radiographic protection gloves need to register their establishment, list the gloves, meet the medical device Quality System regulation, do medical device reporting (MDR), and properly label their gloves. In addition, manufacturers should maintain technical data to show that their attenuation claims are met for the energy range of x-rays normally used in medical procedures.
Embalming gloves are not regulated by the FDA.
Gloves used for food handling or preparation are not medical devices; instead, they are considered by FDA to be a food contact surface which may result in indirect food additives to the food handled. Title 21 CFR 177.2600, Rubber Articles Intended for Repeated Use, lists elastomers, vulcanizing agents, accelerators, activators, coloring agents, etc., and the maximum percentages of these compounds that are permitted by FDA for use in compounding gloves for food contact.
For further information regarding additives and food use of gloves contact the:
Food and Drug Administration
Center for Food Safety and Applied Nutrition
Division of Food and Color Additives (HFF-330)
Harvey W. Wiley Federal Building
Room 1B-018
5100 Paint Branch Parkway
College Park, MD 20740-3835301-436-2335 (phone)
301-436-2764 or 301-436-2798 (fax)
Gloves that are used for routine janitorial functions in medical facilities are not regulated by FDA. However, gloves that are used for cleaning patients, or cleaning or handling surfaces or items contaminated with patient waste or fluids, are medical gloves and must meet the requirements for patient examination gloves.
Non-medical gloves, commonly known as utility, industrial, or general purpose gloves, are used for tasks that do not involve contact with patients or body fluids. Therefore, they are not regulated by the FDA.
It is illegal for manufacturers to relabel non-medical gloves for medical use or to imply in their labeling that such gloves are suitable for medical use. Companies whose names include a medical term should clearly label their industrial gloves, "For non-medical use." Be careful that the labeling does not reflect a medical logo or vignette that implies medical use.
MANUFACTURER NAME IMPLIES MEDICAL DEVICE
As mentioned above, manufacturers of household, food handling and other industrial gloves that have a medical term in their company name are requested to label their industrial gloves, "Not for medical use." Such labeling will help prevent the purchase and use of industrial gloves for medical applications.
Leak detectors are chemical, electromechanical, or electronic systems designed for glove users to monitor the integrity of the glove barrier immediately before and during glove use. FDA considers these devices to be accessories to medical gloves. As such, any device labeled or intended for the medical glove user to detect leaks through the glove barrier before or during use is a medical device and requires FDA clearance before marketing. The product code for glove leak detectors or testers is 79LDQ. Leak detectors are Class I devices requiring 510(k) clearance before marketing.
Leak testers and other equipment used during the production of gloves are production equipment--not medical devices. The selection, use, control, maintenance, etc., of production equipment is covered by the QS regulation in 21 CFR §§820.70 and 820.72.
RELEASE
AGENTS
POWDERED
GLOVES
CONTENT AND FORMAT OF
PMAS FOR ABSORBABLE DUSTING POWDER FOR SURGICAL GLOVES
FIRMS WITH NDA
or PMA for U.S.P. ABSORBABLE DUSTING POWDER
FEDERAL REGISTER,
VOL. 36, NO. 101 - TUESDAY, MAY 25, 1971
During processing, the "outside" of the gloves are coated with a donning lubricant. In most glove manufacturing processes, gloves are inverted when they are stripped from the formers. For most powdered gloves, the "outside" lubricant is cornstarch which remains, after stripping, inside the inverted gloves as the donning lubricant or powder.
Donning lubricants such as cornstarch, silicone, etc., are used to ease insertion of the user’s hand into a glove. Powdered lubricants are also called donning powders or dusting powders. Cornstarch which meets the specification for absorbable donning or dusting powder in the United States Pharmacopeia (U.S.P.) is a commonly used lubricant for patient examination gloves.
Powder used for lubricating examination gloves should meet the U.S.P. monograph for absorbable dusting powder or be shown to be equivalent in terms of safety and effectiveness. The U.S.P. NF XVII Monograph for Absorbable Dusting Powder is presented near the end of this chapter. The 510(k) submission must state the type, specifications and source of powder or other donning lubricant used on the gloves. Talc, cotton flock, and other non-absorbable materials are not acceptable as a lubricating, dusting or donning powder. Lycopodium (club moss spores) and ground pine pollen are toxic and are not acceptable as powder on or in medical gloves. Also, the ASTM standards require that the inside and outside surface of medical gloves be free of talc (paragraph 4.3 of D 3578-91 and D 5250-92, and paragraph 5.3 of D 3577). The ASTM standard for finger cots (paragraph 5.3, D 3772) requires that cots and any dressing materials applied to them not liberate substances known to be toxic or otherwise harmful under normal conditions of use.
Absorbable dusting powder for lubricating a surgeon’s glove is a transitional device (a device formerly regulated as a new drug before l976) and is listed under 21 CFR 878.4480 as a Class III device which requires an approved Premarket Approval Application (PMA) or prior to May 28, l976, a New Drug Application (NDA). Only absorbable dusting powders from powder manufacturers that have an approved PMA or NDA may be used on powdered surgeon’s gloves.
Powder from medical gloves directly contacts wounds, body cavities and skin, and it contaminates the user environment. Due to the enormous numbers of medical gloves used in healthcare, the amount of powder on finished gloves needs to be minimized. To meet QS requirements for device specifications in §§820.30 and 820.181, manufacturers should establish a specification for the amount of powder on a glove. (Also see the labeling information in chapter 6 regarding the powder level on gloves.) The manufacturer should also establish a procedure to verify that the powder level on the finished gloves meet their specification.
CONTENT AND FORMAT OF PMAS FOR ABSORBABLE DUSTING POWDER FOR SURGICAL GLOVES
For information about premarket approval application (PMA) for absorbable powder for lubricating a surgeon's glove, please see the guidance entitled "Premarket Approval Applications (PMA) for Absorbable Powder for Lubricating a Surgeon's Glove at http://www.fda.gov/cdrh/ode/guidance/1230.html
ABSORBABLE DUSTING POWDER, USP
Absorbable Dusting Powder is a specially processed cornstarch. It is a substance recognized in the United States Pharmacopeia - National Formulary (USP-NF). The USP-NF is a standards setting body in the United States. The USP-NF is officially recognized in the Federal Food, Drug and Cosmetic Act (Act).
Under section 502(g) of the Act, if a product is claimed to be the same as one named in an official compendium, including the USP-NF, it must be packaged and labeled in accordance with the requirements stated in the compendium. Failure to meet this requirement causes the product to be misbranded.
Therefore, if you choose to use Absorbable Dusting Powder, USP as a donning powder or glove lubricant, the powder you use must meet the requirements stated in the current revision of the USP-NF. You can get information about obtaining a copy of the current monograph for Absorbable Dusting Powder, USP from the USP Internet web site at: http://www.usp.org/
STERILIZATION OF POWDERED GLOVES:
In addition, validation data, such as the Sterility Assurance Level (SAL) and the organism used as a biological indicator, should be provided, and the validation method for each sterilization process should be described. Since powder is sold nonsterile and is sterilized with gloves by glove manufacturer, then it is the responsibility of the glove manufacturer to validate the sterilization method.
Since surgical gloves may be labeled for resterilization if the package integrity is breached, data on the number of sterilization cycles that the powder can withstand and still remain within specifications should be provided. This is not needed since gloves should be discarded and not resterilized if the package integrity is compromised.
If sterilization with ethylene oxide is specified, then the maximum levels of residues of ethylene oxide, ethylene chlorohydrin, and ethylene glycol which may remain associated with the powder should be provided. The levels should be consistent with the draft Federal Register Notice on ethylene oxide limits. This is not needed since this is the responsibility of the glove manufacturer with the final product.
FIRMS WITH NDA or PMA for U.S.P. ABSORBABLE DUSTING POWDER
(These are companies that have approvals as of February, 1998. This list will be updated as future applications are approved and this manual is reprinted.)
| COMPANY | NUMBER | TRADE NAME |
| Roquette America 1417 Exchange Pl. Keokuk, Iowa 52632 FAX 908-685-5005 |
N83023 N85356 | KEOFLO 7136 KEOFLO 7136p |
| National Starch and Chem. P.O. Box 6500 10 Finderne Ave Bridgewater, New Jersey 08807-0500 FAX 908-685-5005 |
N80535 | ABSORBO - cross linked with oxychloride. ABSORBO-HP-cross linked with epichlorohydrate |
| Grain Processing Corp. 1600 Oregon Street Muscatine, Iowa 52761 FAX 319-264-4495 |
P890070 | PURE-DENT b851 |
| Agrana Starkegesellschaft m.b.H. Conrathstrabe 7 A-3953 GMUEND AUSTRIA FAX 43 2852 503 360, 361 |
P880089 | AGENASORB 9020 |
| A.E. Staley Manufacturing Route #4 P.O. Box 55 Houlton, ME 04730 FAX 207-532-2572 |
P900016 | MIR-FLO Starch |
FEDERAL REGISTER, VOL. 36, NO. 101 - TUESDAY, MAY 25, 1971
DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE
Food and Drug Administration
[DESI 6264]
ABSORBABLE
DUSTING POWDER
Drugs for Human Use; Drug Efficacy Study
Implementation
The Food and Drug Administration has evaluated a report received from the National Academy of Sciences - National Research Council, Drug Efficacy Study Group, on the following drug for use as a glove dusting powder:
Bio-Sorb: nonpeptizable homogeneous mixture of amylose and amylopectine derived from cornstarch with 2 percent magnesium oxide, marketed by Ethicon, Inc., Division of Johnson and Johnson, U.S. Highway 22, Somerville, New Jersey 08876 (NDA 6-264).
Such drugs and similar drugs are regarded as new drugs [21 U.S.C. 321(p)]. Supplemental new drug applications are required to revise the labeling in and to update previously approved applications providing for such drugs. A new drug application is required from any person marketing such drug without approval.
A. Effectiveness classification. The Food and Drug Administration has considered the Academy report, as well as other available evidence, and concludes that a nonpeptizable homogeneous mixture of amylose and amylopectine derived from cornstarch with 2 percent magnesium oxide is effective for use as a biologically absorbable glove powder.
B. Conditions for approval and marketing. The Food and Drug Administration is prepared to approve abbreviated new drug applications and abbreviated supplements to previously approved new drug applications under conditions described herein.
a. The label and other labeling bear the statements:
(1) "Caution: Powder should be removed from the gloves after donning by wiping gloves thoroughly with a sterile wet sponge, sterile wet towel, or other effective method."
(2) Surgical gloves treated with this powder are required to be labeled with the statement: "Caution: After donning, remove powder by wiping gloves thoroughly with a sterile wet sponge, sterile wet towel, or other effective method."
b. The package labeling includes appropriate material which is recommended for display at the point of use and is designed to convey the above cautions to users of the drug or gloves treated with the drug.
c. The drug is labeled to comply with all requirements of the Act and regulations. Its labeling bears adequate information for safe and effective use of the drug. The recommended use of the drug as stated on the label and in any other labeling is as follows: "A biologically absorbable glove powder."
a. For holders of "deemed approved" new drug applications (i.e., an application which became effective on the basis of safety prior to October 10, 1962), the submission of a supplement for revised labeling and an abbreviated supplement for updating information as described in paragraph (n)(1) (i) and (iii) of the notice of July 14, 1970.
b. For any person who does not hold an approved or effective new drug application, the submission of an abbreviated new drug application as described in paragraph (a)(3)(i) of that notice.
c. For any distributor of the drug, the use of labeling in accord with this announcement for any such drug shipped within the jurisdiction of the Act as described in paragraph (b) of the notice.
A copy of the NAS-NRC report has been furnished to the firm referred to above. (this is now out of print)
Communications forwarded in response to this announcement should be identified with the reference number DESI 6264, directed to the attention of the appropriate office listed below and addressed to the Food and Drug Administration, 5600 Fishers Lane, Rockville, Maryland 20857:
Supplements (identify with NDA number):
Office of Scientific Evaluation (BD 100), Bureau of Drugs.
Original abbreviated new drug applications (identify as such):
Drug Efficacy Study Implementation Project Office (BD-5), Bureau of Drugs.
All other communications regarding this announcement:
Drug Efficacy Study Implementation, Project Office (BD-5), Bureau of Drugs.This notice is issued pursuant to the provisions of the Federal Food, Drug, and Cosmetic Act (Secs. 502, 505, 52 Stat. 1050-53 as amended: 21 U.S.C. 352, 355) and under the authority delegated to the Commissioner of Food and Drugs (21 CFR 2.120).
Dated: May 10, 1971
|
SAM D. FINE |
[FR Dec. 71 7218 Filed 5-21-71:8:46 am]
INTRODUCTION
SKIN IRRITATION AND
DERMAL SENSITIZATION STUDIES
Primary Skin Irritation Test (Animal Study)
Dermal Sensitization Study (Animal Study)
Hypoallergenicity
Testing for Skin Sensitization to Chemicals
COLOR AND
FLAVOR ADDITIVES
NON-PYROGENIC
LABORATORIES THAT
PERFORM ASTM DRAIZE TEST
PRIMARY SKIN
IRRITATION AND HUMAN DERMAL TOXICITY TEST LABS
REFERENCE TO ASTM
TEST LAB DIRECTORY
All medical devices, including patient examination and surgical gloves, must be safe and effective for the intended use. Therefore, devices such as gloves that contact the body must be biocompatible.
Biocompatibility data should be submitted for all medical gloves, including synthetic polymer gloves of all types and synthetic polymer-coated latex gloves, latex surgical gloves, and latex patient examination gloves. Because medical gloves are in direct contact with skin, a primary skin irritation study and a dermal sensitization study are appropriate. (See FDA, CDRH, ODE Blue Book Memorandum #G95-1 and ISO TC 10993 for further guidance.) Blue Book Memorandums are available from DSMICA by phoning Facts-On-Demand at 30l-827-0111 or 800-899-0381.
ISO and most other standards are available from the American National Standards Institute (ANSI). 11 West 42nd Street, New York, New York 10036, Phone 212-642-4900, FAX 212-398-0023, http://www.ansi.org/
Each 510(k) application should include the required biocompatibility test results and other information and data as described in this manual. FDA will not review an incomplete 510(k) application. A premarket notification [510(k)] application should contain an attached report for each major study or test program such as biocompatibility studies. All reports or attachments should be identified with the manufacturer’s name, the pages should be numbered, and listed in the table of contents. Biocompatibility tests should be performed on finished gloves. For sterile devices, test data should include the results of tests performed using the finished sterilized devices. (Of course, biocompatibility tests may also be performed on raw materials in order to select qualified materials for use in the devices.)
To facilitate FDA review of the data, analysis, and conclusions in the application, the manufacturer and contract laboratory, if used, should check the:
The summary of test results should be presented in a table format in each report whenever possible. Each study or test attachment report should contain sufficient and well-organized information in reasonable detail so that the FDA reviewer can determine:
A description of the tests and the results obtained are essential; and reasonable and sufficient details of all test procedures and results should be submitted to FDA. For biocompatibility studies, manufacturers should use a standard scoring system for each test method, if a standard scoring system exists. Each test report should include the following:
When a study such as a biocompatibility study is conducted by an internal or contract laboratory to establish a company device specification and/or to obtain data for a submission to FDA, the device manufacturer should keep the original records of the study, as listed above, on file as part of their design verification records in the design history file (DHF). Do not submit the original records to FDA. During factory inspections, FDA investigators may ask to see these original records. For surgeon's gloves and for examination gloves if they become class II, these records are covered by §§820.3(i), 820.30(f) and 820.30(j).
SKIN IRRITATION AND DERMAL SENSITIZATION STUDIES
Anyone wishing to obtain clearance from FDA to market medical gloves including surgical and examination gloves in the United States should supply FDA with data from a Primary Skin Irritation Study and a Dermal Sensitization Study. The gloves used for biocompatibility studies should be finished gloves. That is, the gloves should contain the same colorants, fragrances, powders, lubricants, processing chemicals, etc., and be processed, packaged and, if appropriate, sterilized by the same methods as the gloves to be distributed. The need to repeat biocompatibility studies should be considered if subsequent changes are made in glove composition, manufacturing materials, or processing.
The following is a general discussion of how these skin irritation and dermal sensitization studies may be conducted. A list of laboratories that promote their ability to conduct these tests is printed at the end of this chapter. The list may not be inclusive of all laboratories capable of providing this service and does not constitute an endorsement of these laboratories by FDA. Because methods may vary from laboratory to laboratory, the test data submitted to FDA should contain a brief description of the test protocol, scoring criteria used, and the method used for rating skin responses.
Primary Skin Irritation Test (Animal Study)
Skin irritation testing is performed to demonstrate the irritation potential of the gloves, i.e., for initiating or aggravating damage through its contact with the skin. Primary skin irritation testing is usually done according to the regulations of the Consumer Product Safety Commission, located at 16 CFR Part 1500. The purpose of the study is to determine the dermal irritation potential of the test article to intact and abraded skin of the rabbit.
The backs of six healthy albino rabbits are clipped free of hair. The skin is abraded in one area and left intact in the other. At least a 1 inch x 1 inch portion of the test article (piece of medical glove) is applied to each of two sites per rabbit. The inside and outside of the gloves should be identified such that both sides are tested, i.e., approximately half of the test articles expose opposite sides of the glove to the subject. The test article is covered by a double layer of surgical gauze. The gauze is covered with non-reactive adhesive tape and the entire test site is wrapped with an impervious cloth. The rabbits are returned to their cages.
The condition of the skin is then evaluated after 24 hours of exposure and again at 72 hours. The reactions should be scored according to the skin reaction values as stated under 16 CFR 1500.3(c)(4).
Dermal Sensitization Study (Animal Study)
Dermal sensitization is performed to demonstrate the potential of the device for eliciting a delayed hypersensitivity (Type IV) immunological response through its contact with the skin. This reaction is due primarily to substances which could leach out of a material. Guinea pigs are used because they have been shown to be the best animal model for human allergic contact dermatitis. Methodology for the study is illustrated under ASTM standard F-720-86, Standard Practice for Testing Guinea Pigs For Contact Allergens, Guinea Pig Maximization Test. Laboratories may also use the method of Buehler, as reported in Archives of Dermatology (1965). Dermal sensitization studies use 2 tests or phases: the induction phase and challenge phase. The inside and outside of the gloves should be identified such that both sides are tested, i.e., approximately half of the test articles expose opposite sides of the glove to the subject.
Induction Phase. In Buehler’s method the hair is clipped from the mid-back area of 10 guinea pigs designated as test animals. At least a 1 inch x 1 inch sample of the test article, backed by at least a 1 inch x 1 inch gauze pad, is applied to the test area. The gauze pad is covered with non-reactive adhesive tape and wrapped with an elastic bandage.
Test articles are removed after 6 hours and observations for erythema and edema are recorded. The test article application procedure is repeated 3 times each week for 3 weeks until 9 applications are made to the test area.
Challenge Phase. Two weeks after application of the final induction test article, the hair of each guinea pig, including 5 additional untreated animals used as negative controls, is removed with a clipper from the mid-back area. At least a 1 inch x 1 inch piece of test article is applied to the shaved area of the test and control guinea pigs and taped in place. The trunk of each animal is wrapped with an elastic bandage to maintain the test article on the site. The test articles are removed after 6 hours. Then three observations for erythema and edema are made:
Title 21 CFR 801.437 prohibits the use of the word "hypoallergenicity" on user labeling for natural rubber latex gloves distributed after September 30, l998. This includes gloves that have received prior 510(k) marketing clearance.
FDA does not currently require a new 510(k) submission for labeling changes made to comply with 21 CFR 801.437, provided that no other changes requiring a new 510(k) submission are made to the same device. However, the firm must keep appropriate records documenting the labeling changes.
Testing for Skin Sensitization to Chemicals
Please refer to the document titled, "Draft Guidance on the Content and Format of Premarket Notification [510(k)] Submissions for Testing for Skin Sensitization to Chemicals in Latex Products," available on the World Wide Web at: http://www.fda.gov/cdrh/ode/944.html.
If a manufacturer has already conducted a Modified Draize (MDT) test on a minimum of 200 human subjects to support a reduced sensitization claim, the MDT data may be used instead of the primary skin irritation test (animal) and dermal sensitization study (animal) typically used to support the general biocompatibility of medical gloves.
Manufacturers have the responsibility to demonstrate that color and flavor additives remaining in and on gloves are safe.
Color additive regulations are located in 21 CFR Parts 70 to 82 and flavor additive regulations are located in 21 CFR Part 172 Subpart F. These regulations define acceptable flavor and color additives. The addition of color, flavor, or any chemical to a medical glove is considered to be a significant change which requires a premarket notification submission [510(k)]. Also, a 510(k) submission for a new glove or for a modification to an existing glove may be submitted according to the guidance titled, The New 510(k) Paradigm - Alternative Approaches to Demonstrating Substantial Equivalence in Premarket Notifications, available on the World Wide Web at: http://www.fda.gov/cdrh/ode/parad510.pdf
The Medical Device Amendments of 1976 amended Section 721 of the Food, Drug, and Cosmetic (FD&C) Act to make color additives used in medical devices subject to the same provisions that apply to color additives in food, drugs and cosmetics. The FD&C Act provides that devices containing color additives are to be considered adulterated unless there is a regulation in effect that lists the color for such use. A so-called listing regulation identifies a color and prescribes a specific use. However, the FD&C Act limits the applicability of these provisions to only those color additives that are associated with devices that come in direct contact with the body for a significant period of time. The color additives listed in Subpart D or Parts 73 and 74 of 21 CFR belong to this category.
The color additive regulations for medical devices (21 CFR Parts 73 and 74) should not be confused with the general 510(k) requirements which are independent of the color additive listing and certification mentioned above. It is the manufacturer’s responsibility to show that any substance added to a device, not necessarily limited to color additives, does not adversely affect the safety of the device. Therefore, if a device contains a color or other chemical additive and the device is intended to be in contact with the skin or other parts of the body, various biocompatibility data should be submitted to demonstrate the safety of the additives unless the manufacturer can establish that the additives would not leach and contact the body.
Medical device labeling requirements do not require on the glove box or carton an "ingredient statement" listing the flavor agent, colorant or other additives used in the manufacture of the glove.
FDA does not believe that there is any medical basis for a non-pyrogenic claim for medical gloves, including surgeon's gloves.
The following list was compiled as an aid to the medical device industry. An attempt was made to compile an inclusive list from available public information sources. Inclusion of the name of a manufacturer on this list does not connote FDA recognition of their ability to adequately perform the services listed. As with any supplier of raw materials or services, it is the manufacturer’s responsibility to determine and verify the adequacy of the services offered. See Chapter 10 and 21 CFR 820.30 and 820.50.
LABORATORIES THAT PERFORM ASTM DRAIZE TEST
| Consumer Product Testing
Co. 12 Spielman Road Fairfield, N.J. 07004 |
Ph: 201-226-6146 FAX: 201-808-7234 | ||
| Hilltop Pharmatest,
Inc. 3333 Vine Street Cincinnati, Ohio 45220 |
Ph: 513-281-2989 FAX: 513-281-0148 | ||
| California Skin Research Institute
(CSRI) 15222-B Avenue of Science San Diego, CA 92128
|
Ph: 619-618-1328 Toll Free 800-808-2774 FAX: 619-618-1476 http://www.calskin.com/ | ||
| Lab specialties: Predictive patch studies for medical devices; modified Draize-95 tests on human subjects | |||
NOTE: To add your lab to this Draize test list, please supply formatted data as shown above, including your Email address, during the comment period.
The following list was compiled as an aid to the medical device industry. An attempt was made to compile an inclusive list from available public information sources. Inclusion of the name of a manufacturer on this list does not connote FDA recognition of their ability to adequately perform the services listed. As with any supplier of raw materials or services, it is the manufacturer’s responsibility to determine and verify the adequacy of the services offered. See Chapter 10 and 21 CFR 820.30 and 820.50.
PRIMARY SKIN IRRITATION AND HUMAN DERMAL TOXICITY TEST LABS
| AMA Laboratories, Inc. 216 Congers Road, Bldg. 1 New City, NY 10956
|
Ph: 914-634-4300 FAX: 914-638-4872 | ||
| Lab specialties: Human clinical testing only, modified Draize, 1 climate. | |||
| California Skin Research Institute
(CSRI) 15222-B Avenue of Science San Diego, CA 92128
|
Ph: 619-618-1328 Toll Free 800-808-2774 FAX: 619-618-1476 http://www.calskin.com/ | ||
| Lab specialties: Predictive patch studies for medical devices; modified Draize-95 tests on human subjects | |||
| Clinical Research Laboratories,
Inc. 371 Hoes Lane Piscataway, NJ 08854 USA
|
Ph: 908-981-1616 FAX 908-981-0520 | ||
| Lab specialties: Modified Draize, 1 climate | |||
| Concordia Research Laboratories,
Inc. 248 Columbia Turnpike Florham Park, NJ 07932 USA
|
Ph: 201-734-0734 FAX: 201-734-0334 | ||
| Lab specialties: Human patch testing, oral product research and evaluation, medical device evaluation (safety/efficacy/claim support); modified Draize, 1 climate. | |||
| Consumer Product Testing Company,
Inc. 12 Spielnan Rd Fairfield, NJ 07004 USA
|
Ph: 201-808-7111 FAX: 201-808-7234 | ||
| Lab specialties: Pre-clinical safety and efficacy testing, human patch testing. | |||
| Contox, Ltd. P.O. Box 368 Ft. Washington, PA 19034-0368
|
Ph: 610-277-2458 and 215-288-4882 FAX: 610-277-3980 | ||
| Lab Specialties: Irritation (e.g., 21-day cumulative irritation), allergy (e.g., RIPT), phototoxicity and photoallergy, OTC Monographs, etc.; modified Draize. Human testing only. | |||
| Covance Laboratories 3301 Kinsman Boulevard Madison, WI 53704 USA
|
Ph: 608-242-2622 FAX: 608-241-7227 | ||
| Lab specialties: Clinical testing and research laboratory, safety and efficacy testing, sensory evaluation, patch testing, consumer use studies. No modified Draize studies. | |||
| Education & Research
Foundation 2602 Langhorne Road Lynchburg, VA 24501 USA
|
Ph: 804-847-5695 FAX: 804-846-1707 | ||
| Lab specialties: Dermatology
efficacy studies, patch, photopatch; modified Draize, 1
climate Comments: Main site in Lynchburg; alternate site in Richmond, VA. | |||
| Essex Testing Clinic,
Inc. 799 Bloomfield Avenue, Suite 212 Verona, NJ 07044 USA
|
Ph: 201-857-9541 FAX: 201-857-9662 | ||
| Lab specialties: Human patch testing, all aspects of human safety/efficacy testing for cosmetics, drugs, etc.; modified Draize, 1 climate. | |||
| Harrison Research Laboratories,
Inc. 2497 Vauxhall Rd email: HRLabs@aol.com Union, NJ 07083 USA
|
Ph: 908-688-7600 FAX: 908-688-7601 | ||
| Lab specialties: Human patch testing, efficacy/exaggerated-use tests, claim support; modified Drize, 1 climate. | |||
| Hill Top Research,
Inc. P.O. Box 429501 Cincinnati, OH 45242 USA
|
Ph: 513-831-3114 FAX: 513-831-1217 | ||
| Lab specialties: Human dermal
studies for OTC and RX drugs and personal care products, acute toxicology,
sensory evaluation; modified Draize, 2 climates Comments: Human studies are offered in Cincinnati, Ohio; St. Petersburg, Florida; West Palm Beach, Florida, Scottsdale, Arizona; East Brunswick, NJ; and Winnipeg, Manitoba, Canada. | |||
| Industrial Toxicology Research
Centre Mahatma Gandhi Marg P.B.No. 80 Lucknow - 226 001 INDIA |
FAX: 522-248227 | ||
| Ivy Laboratories,
Inc. University City Science Center 3401 Market Street, Suite 226 Philadelphia, PA 19104 USA
|
Ph: 215-387-8400 FAX: 215-387-1046 | ||
| Lab specialties: Human safety and efficacy patch studies (maximization, irritation, phototoxicity, photoallergenicity). Human testing only. | |||
| MacWill Research
Laboratories 564 Lee Street, S.W. Atlanta, GA 30310 USA
|
Ph: 404-753-1226 FAX: 404-753-9599 | ||
| Lab specialties: Skin irritation and sensitization; modified Draize,1 climate | |||
| NeuroCommunications Research
Laboratories, Inc. Vespucci Drive Ph: 800-336-1935 Danbury, CT 06180 USA
|
Ph: 203-744-7474 FAX: 203-744-7488 | ||
| North American Science Associates,
Inc. 2261 Tracy Road Northwood, OH 43619-1397 USA
|
Ph: 419-666-9455 FAX: 419-666-2954 | ||
| Lab specialties: Primary skin
irritation; non clinical work only Other locations: Kennesaw, Georgia; Irvine California | |||
| Organon Research Centre 7, Wood Street Calcutta - 700 016 INDIA |
FAX: 33-2473750 | ||
| Paddington Testing Company,
Inc. 1819 J.F. Kennedy Boulevard Philadelphia, PA 19103 USA
|
Ph: 215-563-7330 FAX: 215-563-3044 | ||
| Lab specialties: Human patch
testing, cumulative irritancy, clinical trials and acceptance studies;
modified Draize; 2 climates. Two labs in the U.S. plus cooperating
laboratories in Europe, Asia, Africa, and South America.
Pharmaceutical and Cosmetic Evaluations (PACE) Division Lab specialties: Sensory evaluations, damaged or irritated skin evaluations; modified Draize, 2 climates. | |||
| Product Safety Labs A Division of Nutrition International 724 Cranbury Road Ph: 800-425-0002 East Brunswick, NJ 08816-3206 USA
|
Ph: 732-254-9200 FAX: 732-254-6736 | ||
| Lab specialties: Rabbit skin irritation and sensitization; modified Draize; 2 climates. | |||
| Herbert V. Shuster, Inc. 5 Hayward Street Quincy, MA 02171 USA
|
Ph: 617-328-7600 FAX: 617-770-0957 | ||
| Lab specialties:
Performance/efficacy testing, human studies, patch testing; modified
Draize. Comments: Additional facility located in Atlanta, Georgia. | |||
| STS, Inc. P.O. Box 349 7500 West Henrietta Rd. Rush, NY 14543 |
Ph: 716-533-1672 FAX: 716-533-1796 | ||
| West Coast Analytical 9840 Alburtis Santa Fe Springs, CA 90670
|
Ph: 562-948-2225 FAX: 562-948-5850 | ||
| Lab specialty: Analytical testing only | |||
| PPD Pharmaco
International Townfield House, 30-33 Townfield Street Chelmsford, Essex, CM1 1QL England
|
Ph: 44(0) 1245-252878 FAX: 44(0) 1245-490451 | ||
| Lab specialties: Patch testing
(acute, RIPT, maximization, etc.). Comments: Other locations: Cambridge, UK; Chelmsford, UK; Leicester, UK; Southampton, UK; Stockholm, Sweden; Brussels, Belgium; Warsaw, Poland; Karlsruhe, Germany; Gentilly, France; Madrid, Spain; Sidney, Australia; Prague, Czech Republic; Johannesburg, South Africa. U.S. Locations: Austin, Texas; Richmond, Virginia; Columbia, Maryland; Arlington, Virginia; Research Triangle Park, North Carolina; Princeton, New Jersey. | |||
| Haffkine Institute for Training,
Research & Testing Acharya Dandi Marg, Parel, Bombay - 400 012 INDIA |
|||
| Inveresk Research International
Ltd. Corporate Headquarters Tranent EH33 2 NE Scotland
|
Ph: 44(0)1875 614545 FAX: 44(0)1875 614555 | ||
| Lab specialties: Product safety -- skin irritation/delayed contact allergy potential offices in Tokyo, Japan; Frankfurt, Germany; Paris, France; and Falls Church, Virginia, USA. | |||
| Huntingdon Life Sciences, Ltd.
Woolley Road Alconbury Huntingdon Cambs. PE17 5HS England |
Ph 44(0) 1480 892 000 FAX: 44(0) 1480 892 205 | ||
| Lab specialties: Long & short
term toxicology, metabolic studies, environmental studies, pharmacology,
reproductive & mutagenicity studies; no modified Draize
studies Labs located in Huntington, Cambridgeshire; Eye, Suffolk; Wilmslow, Cheshire; and Princeton, New Jersey, USA | |||
| SGS U.S. Testing Company,
Inc. 291 Fairfield Avenue Fairfield, NJ 07004
|
Ph: 973-575-5252 FAX: 973-244-1823 | ||
| Lab specialties: Biocompatibility tests--primary eye and skin irritation, skin sensitization, intracutaneous and systemic toxicity texts, hemolysis; select panel, human patch tests, irritation and sensitivity; cytotoxicity. Protein assay (modified Lowry assay for soluble protein in latex); physical/chemical tests; performance tests, compatibility testing vs. lotions, germicides used in health settings. | |||
| Shriram Institute for Industrial
Research 19, University Road Delhi-110007 INDIA
|
Ph: 91-11-725-7267, 725-7860 FAX: 91-11-725-7676 | ||
| Lab specialties: modified Draize, 2 climates | |||
NOTE: To add your lab to this toxicity test list, please supply formatted data as shown above, including your Email address, during the comment period.
Reference to ASTM Test Lab Directory
| This directory is listed as an aid to the medical device industry. Inclusion of the name of a company does not connote FDA recognition of their ability to adequately perform the services listed. As with any supplier of services, it is the manufacturer’s responsibility to determine and verify the adequacy of the services offered. See 21 CFR 820.30 and 820.50. |
The Association for Testing and Materials (ASTM) has a directory, the International Directory of Testing Laboratories, which lists the locations and capabilities of testing laboratories that perform services for a fee. The information on the types of tests performed, materials analyzed, or other services offered is based on questionnaires signed and submitted by officers of the laboratories. Each laboratory pays a fee for the annual listing.
Starting with the 1988 edition, this Directory became an annual ASTM publication. The 1997 edition includes these features:
The Directory contains two sections-laboratory listings and indexes. The laboratory listings appear geographically so that you can easily scan and select the one or more laboratories in your geographic area that handle the product or service required. Each laboratory has an assigned laboratory number. The indexes in the Directory use this assigned laboratory number when referring to the specified laboratory.
Further information on the services of listed laboratories should be obtained directly from the listees. Direct any questions on using the Directory or including your laboratory’s services in the 1998 edition to: Judy Helm, Marketing Department, ASTM, 100 Barr Harbor Drive, West Conshohocken, PA 19428-2959 (phone 610-832-9610). Inquiries about purchasing this Directory or other ASTM publications should be directed to ASTM Customer Service at 610-832-9585.
| ASTM has not attempted to investigate, rate, endorse, or place a seal of approval upon any laboratory. Nor does ASTM vouch for the qualification of the individual laboratories. Therefore, this Directory is not intended, and should not be used, as an ASTM certified laboratory list of laboratories offering their services for either government or private work. |
Name and Place of Business
Statement of Identity
Net Quantity of Contents Statement
Country of Origin
Adequate Directions for Use
Powder and Protein Labeling
Expiration Date
Lot Number
Donning Powder or Lubricant Identification
Standards
Bar Coding
National Health Related Items Code
"Powder-Free"
Protein Label Claims
Polymer-Coated Gloves
Chemical Sensitization
Color and Flavor Additives
Chemotherapy Label Claim
Non-Pyrogenic
Hypoallergenicity
Special Label Claims *
Required and recommended labeling for medical gloves are described below along with examples of typical labeling for dispenser boxes of examination gloves and typical labeling for a unit package containing a pair of surgeon’s gloves.
BASIC LABELING REQUIREMENTS (21 CFR Part 801)
Name and Place of Business (21 CFR 801.1)
Statement of Identity (21 CFR 801.61)
Net Quantity of Contents Statement (21 CFR 801.62)
The label must contain a statement of net quantity of contents in terms of weight, numerical count, or statements of both numerical count and weight. Whichever statement of net quantity of contents is used, it must be clearly and understandably stated on the label; for example, "100 gloves -- packaged by weight."
The declaration shall appear as a separate item in the lower 30 percent of each principal display panel; and be separated by at least a space equal to the height of the lettering used in the declaration, from other information appearing above and below, and separated by at least twice the width of the letter "N" from labeling to the left or right.
The label must contain the country of origin if other than the U.S. This is a U.S. Customs requirement.
Adequate Directions for Use (21 CFR 801.5)
Disposable medical gloves should be labeled "single use only," if a symbol is used. The label for surgeon’s gloves must contain any necessary directions for use. The following statement is required (36 FR 9475, May 25, 1971) for sterile powdered surgeon’s gloves:
"Caution: After donning, remove powder by wiping gloves thoroughly with a sterile wet sponge, sterile wet towel, or other effective method."
A similar caution is recommended for powdered patient examination gloves because patient examination gloves are used in a variety of circumstances, including procedures where the surface of the glove contacts wounds, body cavities, or other possible routes of contamination. If conditions warrant, the user may wish to remove residual powder from the gloves prior to use in order to minimize the potential for adverse effects. For this reason, FDA recommends the following statement appear on each box of powdered patient examination gloves.
"Caution: Users should consider the circumstances of use in deciding whether to remove powder from gloves after donning. Powder can be removed by wiping gloves thoroughly with a sterile wet sponge, sterile wet towel, or other effective method."
Powder and Protein Labeling (Proposed 21 CFR 801.440)
The labeling of gloves would be required to bear identifying statements per proposed 21 CFR §801.440 User labeling for powdered and powder-free surgeon's and patient examination gloves. Powder and protein levels shall be displayed in accordance with the labeling requirements as defined in the special control, Medical Glove Guidance Manual (this manual).
Manufacturers with cleared submissions under section 510(k) of the act for surgeon’s or patient examination gloves are not required to submit new 510(k) submissions for labeling changes to add protein and/or powder levels. However, the manufacturer must keep appropriate records documenting the labeling changes.
The caution statements proposed in §801.440 would be required to appear on all device labels, and other labeling, and shall appear on the principal display panel of the device packaging, the outside package, container or wrapper, and the immediate device package, container, or wrapper. These statements shall be prominently displayed in bold print in conformance with section 502(c) of the act.
Natural rubber latex powdered gloves. For natural rubber latex powdered surgeon’s gloves and powdered patient examination gloves, the statement required in 21 CFR §801.437(d) would be superseded by 21 CFR §801.440(a) to read as follows:
"Caution: This product contains natural rubber latex which may cause allergic reactions. FDA recommends that this product contain not more than 120 mg powder and 1200 µg extractable protein per glove. This product contains no more than [insert level] mg powder and no more than [insert level] µg extractable protein."
Synthetic material powdered gloves. For synthetic material powdered surgeon's or powdered patient examination gloves, the labeling would be required by §801.437(b) to prominently bear the following statement:
"Caution: Glove powder is associated with adverse reactions. FDA recommends that this product contain no more than 120 mg powder per glove. This product contains no more than [insert level] mg powder per glove."
Note that §801.440 would not require additional labeling for powder-free synthetic material gloves.
Natural rubber latex powder-free gloves. For natural rubber latex powder-free surgeon's gloves and powder-free patient examination gloves, the statement required in §801.437(d) of this subchapter would be superseded by §801.440(c) to read as follows:
"Caution: This product contains natural rubber latex which may cause allergic reactions. FDA recommends that this product contain no more than 1200 µg extractable protein per glove. This product contains [insert level] µg extractable protein per glove."
At present, the FDA does not allow a protein labeling statement or claim below the current 50µg/gram of glove sensitivity limit of the ASTM Lowry test method (note that for a 6 gram glove, 50µg/gm translates to 6 x 50 = 300µg per glove). This lower limit for protein labeling may change if the ongoing work on ASTM D 5712 results in a more sensitive test method.
For gloves to be labeled as containing µg/gram or less per glove of extractable protein, the labeling should also state:
"Caution: Safe use of these gloves by latex sensitized individuals has not been established."
Expiration Date [Proposed 21 CFR 801.440(d)]
Since the early 1990s, the FDA has encouraged manufacturers to collect data to substantiate the shelf life (expiration date) of each glove product they manufactured. Proposed 21 CFR §801.440(d) would require that all surgeon’s and patient examination gloves bear an expiration date as reprinted below:
(d) All surgeon’s and patient examination gloves shall bear an expiration date as follows:
(1) The expiration date shall state the month and year of the shelf life as supported by data from the studies described in paragraph (d)(3) of this section;
(2) The expiration date must be prominently displayed on the exterior of the primary and retail package, and on the shipping carton;
(3) The expiration date must be supported by stability studies demonstrating acceptable physical and mechanical integrity of the product over the shelf-life of the product from its date of manufacture;
(4) For each glove design, the testing data and stability study protocol supporting an expiration date must be maintained by the manufacturer for a period equivalent to the design and expected life of that glove type, and shall be made available for inspection and copying by FDA; and
(5) Sterile surgeon’s and patient examination gloves that have a date of expiration based on sterility that is different from the expiration date based upon physical and mechanical integrity testing shall bear only the earlier expiration date.
The expiration date should reflect the month and year, for example: January 2002. It should not be stated as 1/10/02 because this could or would be interpreted as October 1, 2002 in some parts of the world, resulting in the use of outdated and degraded gloves.
In the past, expiration dates were based on real time studies by manufacturers. FDA has drafted guidance that allows manufacturers to make a shelf life claim based on accelerated aging techniques and it will be placed on the CDRH web site. Such claims must be verified by real time studies.
Aging studies are performed with a statistically valid number of representative gloves under specified conditions according to a written protocol. If you plan to have a shelf life of several years, you should consider protecting the dispenser box from moisture and contamination. Your packaging system is important for maximizing the shelf life of your products. Increasing shelf life may require dispenser boxes to be laminated or shrink wrapped with plastic film or other method to reduce exposure to moisture and ozone.
Manufacturers with cleared submissions under section 510(k) of the act for surgeon’s or patient examination gloves are not required to submit new 510(k) submissions to add an expiration date to the labeling.
It is customary for the package of medical gloves to bear a lot number. A lot number should identify the batch of compounded latex, the production lines, the production shift, and, if sterile, the sterilization run. A lot number is required by ASTM standards D 3577 section 10.2, D 3578 section 9.3 or D 5250 section 9.3. The lot number should be visible -- not placed on the inside of a dispenser box.
Donning Powder or Lubricant Identification
If surgeon’s gloves are powdered, they must be powdered with an absorbable dusting powder which has received FDA approval under either an NDA or a PMA, and the labeling should inform users with a statement such as, "Powdered with absorbable dusting powder."
If patient examination gloves are powdered, the powder should meet the United States Pharmacopoeia (U.S.P.) monograph for absorbable dusting powder or be shown to be equivalent in terms of safety and effectiveness. The 510(k) for the gloves must state the type, specifications and source of powder or other donning lubricant used on the gloves. This data must demonstrate the biocompatibility of the powder and its lack of adverse effects upon the physical characteristics of the glove. U.S.P powder is commonly used on examination gloves so the corresponding labeling statement should read, "Powdered with absorbable dusting powder, U.S.P."
FDA does not object to manufacturers stating in their labeling that their product meets a specific national or international consensus standard(s). The labeling should clearly identify the standard by name or alphanumeric text including the year published or other information needed to identify the specific standard. Labeling shall not be confusing or misleading; therefore, FDA expects the product to meet all of the applicable parts or parameters of the standard, if such a claim is made. This is guidance for a labeling claim and thus is different from a premarket submission which may contain a declaration of conformity with any or all parts of a standard without an associated claim in the labeling.]
If the label states that a product meets a specific standard, the product delivered to the customer must meet the standard. Thus, the manufacturer should have design, expiration and/or other valid shelf life data on file to support their claim.
Bar coding is favored in several countries such as Europe, Japan and the United States for quick tracking of distributor or hospital inventory. While not an FDA requirement, purchasers may demand that products they buy contain a bar code on cartons and dispenser boxes. Many manufacturers have started placing bar codes on cartons and on the bottom of dispenser boxes.
National Health Related Items Code
The National Health Related Items Code (NHRIC) is a voluntary identification numbering system for medical devices. Purchasers may request that you provide an NHRIC number on your medical devices. The NHRIC is assigned and administered by the CDRH Office of Compliance. The phone number is 301-827-4555 ext.104. If you choose to place an NHRIC on your labeling, it should be preceded by the letter "H" to distinguish it from NDC or Universal Product Code (UPC) numbers and it should prominently appear in the top third of the principal display panel.
In addition to basic labeling described above, manufacturers may have labeling claims for the attributes of their gloves. The claims should be for characteristics of their gloves that are substantially equivalent to characteristics of predicate gloves or that meet a consensus standard. Some attributes are color, flavor, scent, and thickness. Data must be submitted in a premarket notification to support all claims. Ambiguous labeling claims such as "extra thick" or "super-sensitive" should not be included. However, a factual and definitive statement such as, "Twice the minimum ASTM thickness." is acceptable. Claims may not be false or misleading in any way, nor may the quality of the product fall below that which it purports or is represented to possess.
Gloves with trace amounts of residual former-release and donning powders are commonly referred to as "powder free." Most manufacturers dip the glove mold, commonly referred to as a "former," into a solution of calcium carbonate and calcium nitrate. After controlled drying, the coated former is dipped into the latex solution and a glove is created on the former. The calcium carbonate (powder) helps release the glove from the former. Only a small amount of calcium carbonate remains on the "uncured" glove and most of it is removed by leaching and washing.
The finished gloves are tacky and will stick to the hand; therefore, a lubricant is usually applied to the inner side of the gloves to aid in donning. Some manufacturers use surface chlorination and washing to remove manufacturing former-release or stripping powder and to give the glove a slick texture which precludes the need for donning powder. Other manufacturers remove the former-release or stripping powder from the surface of the gloves and use a non-powder donning lubricant such as silicone. Various manufacturers also use proprietary methods to achieve powder-free gloves.
FDA requires a 510(k) for a powder-free glove or a change from a powdered to a powder-free glove. The manufacturing process for producing a powder free glove should be described in detail in the premarket notification. Information demonstrating that the process used does not have a significant effect on the finished glove specifications should also be included.
ASTM has published a standard method D 6124 for collecting and measuring the manufacturing debris, residual former-release powder, etc., on a powder-free glove.
To establish a "powder-free" claim, FDA recommends no more than 2 mg of residual or trace powder and debris per glove, as determined by the ASTM D 6124 test method or an equivalent method. In order for an applicant to substantiate a "powder-free" claim in their 510(k) submission, the applicant should state whether the manufacturing process for the glove includes any powder such as a former-release powder and/or a donning powder. If it does, the applicant should provide:
If the entire manufacturing process does not include any former release on donning powder, then the applicant need only discuss items 4 and 5 in the above list.
Latex protein is reported as a cause of Type I sensitivity in some individuals who have been exposed to latex containing devices. Repeated exposure to latex protein is believed to increase the probability that an individual will become sensitized. Since May 1991, the FDA has recommended that manufacturers of latex devices reduce the water-extractable protein on their natural rubber latex devices. Such reduction is required by the Quality System Regulation, 21 Code of Federal Regulations, Part 820 by 820.3(p), Manufacturing Material, and 820.70(h), Manufacturing Material. However, in 1991 a labeling claim was not allowed in a 510(k) submission for a "protein content labeling claim" because a standard test method for measuring water-extractable protein in natural rubber latex did not exist. Subsequently, the American Society for Testing and Materials, published the ASTM Standard Test Method for Analysis of Protein in Natural Rubber and its Products, D 5712-95. Afterwards, 510(k) submissions with protein claims were accepted by CDRH.
As noted above, FDA would require in proposed 21 CFR 801.440 that the labeling for all latex medical gloves bear a statement declaring the maximum water soluble protein level per glove as measured by the current ASTM D 5712 modified Lowry method or an equivalent method. The FDA recommended limit for water extractable protein is 1200 µg per glove.
Please note that the FDA does not allow a labeling claim for extractable protein below 300µg per glove because of current limits to the sensitivity of the ASTM D 5712-95 Lowry test method which is 50 µg per gram of glove. For a 6 gram glove, 50 µg per gram of glove translates to 300µg per glove ( 6 grams X 50 µg/gm = 300 µg). This lower labeling limit may change if the ongoing work on ASTM D 5712 results in a more sensitive test method.
The water-extractable protein should be measured on recently manufactured finished gloves that have undergone accelerated aging per ASTM standard D 3577 or D 3578 or real time aging. For the data submitted in your 510(k), we suggest that accelerated aging be done for 7 days at 70° Centigrade or real time aging for 3 months to a year.
| The statement declaring the maximum water soluble protein
level should be based upon the upper process limit for
each glove type as determined by testing. In the example shown graphically
below, the upper process limit is 600µg water extractable protein per
glove as determined by testing. Therefore, that lot of gloves should be
labeled:
"This latex glove contains no more than 600µg extractable protein per glove." Do not label the lot with either the process set point or the average level. This is only an example. Your label statement should reflect actual values from your manufacturing lines. The levels shown in this example are not FDA limits, requirements or recommendations. Note that the same principle should be applied to powder levels. For this example, your manufacturing process set point or center operating point should be significantly less than 600µg per glove such that the set point plus process variations produce gloves that contain no more than 600µg per glove of water-extractable protein as shown in the following chart.
For this example, a portion of the labeling statement required by proposed 21 CFR §801.440 would be made as follows: "This latex glove contains no more than 600µg extractable protein per glove." |
To meet Quality System requirements, the processes used to control water-soluble proteins and manufacturing materials must be developed, validated, documented and, thereafter controlled. Validation is required because the protein and residual chemicals on each glove are not measured.
The labeling changes to dispenser boxes and any changes to manufacturing processes must be done according to Quality Systems change control requirements in §§820.30, 820.40 and 820.70. (However, examination gloves are not required to meet §820.30 until the effective date of the final rule when the reclassification to Class II becomes effective.
If a manufacturer coats their gloves to bind extractable proteins and/or aid in donning, then the manufacturer should perform accelerated or real-time aging tests to show that the coating is effective for the normal expected life of the gloves. FDA is aware of complaints that coatings have flaked off or delaminated before the gloves are used. On occasion, synthetic polymer gloves have been reported to be contaminated with latex proteins. Synthetic polymer gloves, polymer-coated latex gloves and any gloves with a specified protein level should not be exposed to airborne protein-coated starch or dipped in any tank where regular protein containing latex gloves have been processed unless the tanks are thoroughly cleaned before the production of the specified protein or non-protein gloves.
As noted in the Federal Register final rule of September 30, 1997 "Latex-Containing Devices; User Labeling," FDA has developed guidance on chemical sensitization. Please refer to the document entitled, "Draft Guidance on the Content and Format of Premarket Notification [510(k)] Submissions for Testing for Skin Sensitization to Chemicals in Latex Products," available on the World Wide Web at: http://www.fda.gov/cdrh/ode/944.html.
Color or flavor additives added to medical gloves during the manufacturing process require biocompatibility data in the premarket notification submission. The addition of colorants, other than traditional whiteners such as titanium dioxide, or the addition of a flavor to a medical glove is considered to be a significant change which requires a premarket notification submission [510(k)]. (Please see Chapter 5, Biocompatibility.)
Medical device labeling requirements do not require on the glove box or carton an "ingredients statement" listing the flavor agents or colorants used in the manufacture of the gloves.
Chemotherapy gloves are specialty medical examination gloves and require premarket notification [510(k)] clearance from FDA before marketing. Chemotherapy gloves should meet the ASTM standard D 3578 or an equivalent standard for examination gloves; however, they are usually 0.10 mm or more in thickness which is more than the 0.08 mm minimum allowed for examination gloves.
To help assure that the 510(k) application is complete and to help FDA determine that the applicant’s gloves are substantially equivalent to legally marketed chemotherapy gloves, the applicant may use the 510(k) format in Chapter 8 for examination gloves. Labeling, donning powder or lubricant, protein, powder-free, etc., requirements for chemotherapy gloves are the same as for examination gloves. In addition, the applicant should specify the chemicals against which the gloves will provide protection and include data to demonstrate that the chemotherapy or other specialty gloves are safe and effective for handling the chemotherapy agents or other claimed special use. The applicant should include in the 510(k) submission:
To market the glove for use in the handling and/or preparation of chemotherapeutic drugs, the glove should be labeled as an "Examination Glove" and "Tested for use with [name of chemotherapeutic drug(s)].
Recommendations of additional information that should be provided or included in labeling are as follows:
The above statements/instructions would provide the user with the information needed to make an appropriate product selection.
The minimum biocompatibility tests for chemotherapy gloves are skin irritation and dermal sensitization.
FDA does not believe that there is a medical basis for a non-pyrogenic claim for medical gloves, including surgeon's gloves.
FDA has received reports of sensitivity to medical gloves labeled as "hypoallergenic." The latex labeling rule, published in the FR on September 30, l997, announced that effective September 30, l998, FDA will not allow the term "hypoallergenic" on the labeling of a natural rubber latex device. FDA believes that this term erroneously implies that the user of products labeled as hypoallergenic is assured that the risk of an allergic reaction to the chemicals or latex proteins in the products would be minimal. In the past, use of the "hypoallergenic" claim has been based on results of the modified (human) Draize test. While this test may be appropriate for detecting sensitivity to residual levels of processing chemicals, the test cannot accurately detect the presence or absence of natural latex proteins. Furthermore, current manufacturing processes cannot reduce the natural latex proteins below the level to which some individuals may be sensitive. Therefore, the FDA believes that the presence of the term "hypoallergenic" on the labeling of a natural rubber latex-containing device is misleading because it incorrectly implies that the product labeled as "hypoallergenic" may be used safely by latex sensitive persons.
The health care community and FDA are interested in improvements in the safety and performance of medical gloves. Some needed improvements are better barrier protection; better resistance to cuts, punctures and tears; longer shelf life and better biocompatibility characteristics. Some of these factors, such as biocompatibility, are addressed by CDRH guidance on protein claims, sensitization tests, irritation tests, etc.
If you wish to make claims not covered by FDA guidance or consensus standards, you should do the following in the order presented below:
FDA Center for Devices and Radiological Health
Infection Control Devices Branch, HFZ-480
9200 Corporate Blvd.
Rockville, MD 20850 USA
Do not send a FAX or email.
LABEL EXHIBITS – The following examples are consistent with proposed 21 CFR 801.440:
Example: POWDERED LATEX PATIENT EXAMINATION GLOVE BOX, TOP
DR# GON®
BRAND
|
CONTENTS: 100 Gloves (by weight)
SIZE:
MEDIUM
Distributed by:
ABC Corporation
Boston, MA
10001
Example: POWDERED LATEX PATIENT EXAMINATION GLOVE BOX, SIDE
DR# GON® BRAND
|
| CONTENTS: 100 Gloves (by weight) Powdered with absorbable dusting powder, U.S.P. |
SIZE: MEDIUM |
Singe Use Only
"Caution: Users should consider the circumstances of use in deciding whether to remove residual powder from gloves after donning. Powder can be removed by wiping gloves thoroughly with a sterile wet sponge, sterile wet towel, or other effective method."
| Distributed by: ABC Corporation Boston, MA 10001 |
Product of Malaysia |
Expires: Aug. 2002 |
Sample: POWDER-FREE LATEX PATIENT EXAMINATION GLOVE BOX
DR# GON® BRAND
|
CONTENTS: 100 Gloves (by weight)
SIZE:
MEDIUM
Manufactured For:
Andywill Care Inc.
Comfort, NC 27777
USA
Sample: POWDER-FREE PATIENT EXAMINATION GLOVE BOX, SIDE
DR# GON® BRAND
|
| Manufactured For: Andywill Care Inc. Comfort, NC 27777 USA |
Product of Thailand |
Expires: Dec. 2002 |
Sample: POWDERED VINYL PATIENT EXAMINATION GLOVE BOX
DR# GON® BRAND
|
CONTENTS: 100 Gloves (by weight)
SIZE:
MEDIUM
Manufactured For:
Medical Art, Inc.
Terrell, MD 28888
USA
Sample: POWDERED VINYL PATIENT EXAMINATION GLOVE BOX, SIDE
DR# GON® BRAND
|
| CONTENTS: 100 Gloves (by weight) |
SIZE: MEDIUM |
Single Use Only
Powdered with absorbable dusting
powder, U.S.P.
"Caution: Users should consider the circumstances of use in deciding whether to remove residual powder from gloves after donning. Powder can be removed by wiping gloves thoroughly with a sterile wet sponge, sterile wet towel, or other effective method."
| Manufactured For: Medical Art, Inc. Terrell, MD 28888 USA |
Product of Taiwan |
Expires: Dec. 2002 |
SAMPLE: POWDERED SURGICAL GLOVE UNIT PACKAGE
|
¯¯ PEEL DOWN TO OPEN ¯¯ DR# GON® BRANDSTERILEPOWDERED LATEX
|
| Lot: S101000 | Expiration Date: Nov. 2002 |
| Distributed by: ABC Corporation Big Apple, NY 10018 |
Product of Indonesia |
INTRODUCTION
TRUTHFUL AND ACCURATE
STATEMENT AND FORMAT
INDICATIONS FOR USE
STATEMENT AND SAMPLE FORMAT
510(k) SUMMARY OR
STATEMENT
Use of Summaries and Statements
Who Responds to Requests for 510(k) Information
REQUIREMENTS
FOR A 510(k) SUMMARY
REQUIREMENTS FOR A
510(k) STATEMENT AND SAMPLE FORMAT
As outlined in Chapter 2 of this manual, a premarket notification 510(k) submission needs to include:
This information helps assure the accuracy of submissions, overtly states the indications for use of the device, speeds the processing of the submission, and supports the fulfillment of requests by the public for information under the Freedom of Information (FOI) Act. These are described below.
TRUTHFUL AND ACCURATE STATEMENT AND FORMAT
All 510(k) submitters must include a statement certifying that all information contained in the 510(k) submission is truthful and accurate and that no material fact has been omitted.
The truthful and accurate statement must be on a separate page and must be identified in the table of contents.
A sample is provided on the next page.
Sample
PREMARKET NOTIFICATION
TRUTHFUL AND ACCURATE STATEMENT
[As required by 21 CFR 807.87(j)]
| I certify that, in my capacity as |
(________________________________________________) |
of |
|
I believe to the best of my knowledge, that all data and information submitted in the premarket notification are truthful and accurate and that no material fact has been omitted.
|
|
|
|
|
|
|
|
The statement must be signed by a responsible person of the company required to submit the premarket notification — not a consultant for the submitter.
* For a new submission, do not fill in the 510(k) number. The Food and Drug Administration will fill in this blank with your 510(k) number when the number is assigned.
INDICATIONS FOR USE STATEMENT AND SAMPLE FORMAT
Each 510(k) submission must include an "Indications for Use" page that contains the applicant’s name, name of the device and the indications for use of the device. For medical gloves, the indication for use is the same as the intended use. The Indications for Use page should contain the 510(k) number for the submission when the 510(k) number is known (for example, when submitting additional information requested by FDA). For a new submission, however, you will not know the number and will not be able to include it. The Indications for Use statement should be located in the front part of your 510(k) submission.. An example of an optional format is on the next page which you may use. It may be completed by using one of the following statements or equivalent text.
The following statements are from the proposed reclassification of medical gloves.
You may use equivalent text that correctly describes the indications for use of your gloves. For a special purpose glove, you must include additional text in the Indications for Use statement that covers the additional function of the glove as claimed in your labeling. The FDA, CDRH, Office of Device Evaluation (ODE) will review the contents of your entire 510(k) submission to determine if the submission supports your Indications for Use statement. Therefore, the information, data and labeling claims in the entire 510(k) submission must support and agree with the Indications for Use statement. The Indications for Use statement will be attached by ODE to the substantial equivalence (SE) letter which ODE will send to the submitter to define for what use the glove was cleared for marketing.
|
|
____________________________________________________________ |
510(k) Number (if known):* |
_____________________________________________________ |
|
|
________________________________________________________________ |
Indications For Use:
*For new submissions, do NOT fill in the 510(k) number.
Section 513(i) of the FD&C Act requires that a person submitting a premarket notification [510(k)] to the FDA must include either:
Use of Summaries and Statements
Summaries are used by FDA to fulfill requests made under the Freedom of Information (FOI) Act. Statements are used to make arrangements for the applicant or certifier to respond to requests.
Who Responds to Requests for 510(k) Information
In instances where a manufacturer or other applicant provides a summary with the 510(k) submission to satisfy the conditions in (1) above, written requests by individuals for copies of the 510(k) summary will be furnished by the FDA through the FOI process after determining that the device is substantially equivalent to another device.
If a manufacturer or other person submitting a 510(k) chooses to provide a statement to satisfy the conditions in (2) above, written requests by any individual for a copy of the 510(k), excluding patient identifiers and trade secret and confidential commercial information, must be fulfilled by the statement certifier within 30 days of receipt of the request. On a monthly basis the FDA publishes the list of names of certifiers of premarket notification submissions for which substantial equivalence determinations have been made [§807.93(b)]. A submitter of a 510(k) may not charge requesters for compiling and disseminating this data.
The choice between the above summary and statement should be made before the 510(k) is submitted. However, submitters may elect to change their choice between a summary or statement before the substantial equivalence determination is reached. After this determination is made, submitters cannot change their choice of a summary or statement.
REQUIREMENTS FOR A 510(k) SUMMARY
If you choose to meet the conditions for a summary, then a summary must be submitted with your 510(k) application and clearly marked as such in order for the FDA to begin its review of a 510(k) submission. A complete and correct summary as described below must be submitted in order for FDA to complete its review of a 510(k) submission. As required by §807.92(a), FDA will accept summaries and amendments thereto until FDA issues a determination of substantial equivalence.
Please make a copy of the following to use as a checklist and check off each item to make sure your summary is adequate and complete.
1 [ ] The summary is a separate section of the submission, beginning on a new page and ending on a page not shared with any other part of the premarket notification submission, and is clearly identified as "510(k) SUMMARY" as required by §807.92(c).
2 [ ] The summary contains on the first page, preferably on your letterhead paper, the submitter’s name, address, phone and Fax numbers, name of contact person, and date the summary was prepared [§807.92(a)(1)].
3 [ ] The summary includes the name of the device, including the trade or proprietary name if applicable, the common or usual name, and the classification name, if known [§807.92(a)(2)].
Examples:
4 [ ] The summary identifies the legally marketed device to which your company is claiming equivalence [§807.92(a)(3)].
Example: Class I* powdered latex patient examination glove 80LYY, powdered
with absorbable dusting powder, that meets all of the requirements of ASTM
standard D 3578-95.
* [Class II if proposal becomes a rule.]
5 [ ] The summary includes a description of the device [§807.92(a)(4)].
For gloves, simply repeat the information in step 4 above and describe any variations. For special purpose gloves describe the special features. For example, for orthopedic gloves, also add as appropriate: The gloves are ## mm thick (and have technical features) to reduce damage from contacting bone, teeth or instruments.
6 [ ] The summary describes the intended use of the device [§807.92(a)(5)].
For gloves, you may select indications for use text from the appropriate one of the 4 numbered proposed paragraphs on preceding page 7-3 that matches your gloves, or equivalent text. This text should agree with the text in your "Indications For Use" statement.
7 [ ] Per §807.92(a)(6), the 510(k) summary contains a summary of the technological characteristics of your device compared to the predicate device. If your device has different technological characteristics from the predicate device, the 510(k) summary contains a summary of how the technological characteristics of your device compare to a legally marketed device to which you are claiming equivalence.
For gloves, include a brief table of: the measured parameters of your finished gloves compared to ASTM or equivalent standards; data that shows compliance with FDA biocompatibility, pinhole, powder-free and other requirements and recommendations; and any other parameter for which you have a labeling claim.
8 [ ] If the determination of substantial equivalence is also based on an assessment of performance data, the summary includes a brief discussion of the nonclinical tests and how their results support a determination of substantial equivalence [§807.92(b)(1)].
Example - The performance test data is the same as for §807.92(a)(6) mentioned immediately above.
9 [ ] If the determination of substantial equivalence is also based on an assessment of performance data, the summary includes a brief discussion of clinical tests and how their results support a determination of substantial equivalence [§807.92(b)(2)].
Clinical data is not needed for gloves or for most devices cleared by the 510(k) process.
10 [ ] Per §807.92(b)(3), the summary includes the conclusions drawn from the nonclinical and clinical tests in (b)(1) and (b)(2). [See steps 8 and 9 above.]
For gloves, state that your gloves:
11 [ ] Per §807.92(d), the summary includes any other standards, special controls, labeling, or regulatory information reasonably deemed necessary by the FDA. Such additional information requested by FDA during review of the submission may include additional safety and effectiveness information and FDA may request that you update your summary.
Please make sure you have included all of the information listed in steps 1 to 11 above and verify that the following criteria have been met.
If you use a summary, writing and reviewing the summary are the last steps in preparing your submission.
After completing your 510(k) according to the format in chapter 8 for examination gloves or chapter 9 for surgeon’s gloves, make two copies of your complete 510(k) including your signed summary. Keep one copy for your records. Submit the complete original 510(k), including the summary, and a complete copy of the 510(k), including the summary, to the FDA.
REQUIREMENTS FOR A 510(k) STATEMENT AND SAMPLE FORMAT
For persons who choose to submit a statement with their 510(k) to the FDA, the specific statement shown below must be submitted with the 510(k) in order for FDA to begin the review process. The statement should be on a separate letterhead page, clearly identified as "510(k) statement," signed by the certifier — not a consultant to the 510(k) submitter, and must include the specific language beginning with "I certify ...," shown in the following sample as required by 21 CFR 807.93:
|
[ your company letterhead ] 510(K) STATEMENT "I certify that in my capacity as ( the position held in company by the person required to submit the premarket notification, preferably the official correspondent ) of ( manufacturer’s name ), I will make available all information included in this premarket notification on safety and effectiveness within 30 days of request by any person if the device described in the premarket notification submission is determined to be substantially equivalent. The information I agree to make available will be a duplicate of the premarket notification submission, including any adverse safety and effectiveness information, but excluding all patient identifiers, and trade secret and confidential commercial information, as defined in 21 CFR 20.61."
|
* For a new submission, do not fill in the 510(k) number. The FDA will fill in this section with your 510(k) number when the number is assigned.
If you use a statement, writing and reviewing the statement are the last steps in preparing your submission.
After completing your 510(k) according to the format in chapter 8 for examination gloves or chapter 9 for surgeon’s gloves, make two copies of your complete 510(k) including your signed statement. Keep one copy for your records. Submit the complete original 510(k), including the statement, and a complete copy of the 510(k), including the statement, to the FDA.
DEFINITION AND
REQUIREMENTS
VOLUNTARY
STANDARDS
BIOBURDEN AND
MOISTURE
PREMARKET NOTIFICATION SUBMISSION FORMAT
New 510(k) Paradigm
Sample Format for A Premarket Notification [510(K)] for Examination Gloves
Premarket Notification 510(k) Submission Applicant
Truthful and Accurate Statement
Indications for Use Statement
Glove Proprietary or Trade Name
Name and Location of ACTUAL Manufacturer
Labels, Labeling, and Advertising
Classification Information
Specifications
Quality Assurance Testing
Sterility
FORMER Release Powder or Chemical
Dusting or Donning Powder
Weight of Powder-free Residue
Protein Level of Natural Rubber Latex Gloves
Protein Control
Chemical Sensitivity Claim
Color or Flavor Additives
Biocompatibility
Expiration Date or Quality at Delivery
Other Claims Requiring Data
510(k) Summary/Statement Requirement: *
Under the proposed rule, patient examination gloves would be Class II (special controls) devices and would be identified as follows:
Patient Examination Gloves, powdered (proposed §880.6250). A powdered patient examination glove is a disposable device made of natural rubber latex or synthetic material that bears powder to facilitate donning and is intended to be worn on the hand or finger(s) for medical purposes to provide a barrier against potentially infectious materials and other contaminants.
The proposed Class II special controls are:
Patient Examination Gloves, powder-free (proposed §880.6251). A powder-free patient examination glove is a disposable device made of natural rubber latex or synthetic material that may bear a trace amount of glove powder and is intended to be worn on the hand or finger(s) for medical purposes to provide a barrier against potentially infectious materials and other contaminants.
The proposed Class II special controls are:
Manufacturers of patient examination gloves are subject to the registration, listing, 510(k), labeling, Quality System and Medical Device Reporting (MDR) requirements of the Food, Drug and Cosmetic Act. Variations in patient examination gloves are listed in Table 3.1 in Chapter 3. Manufacturers must receive a 510(k) clearance letter from FDA before distributing medical gloves in the U.S.
Powder used for lubricating examination gloves should meet the United States Pharmacopoeia (U.S.P.) monograph for absorbable dusting powder or be shown to be equivalent in terms of safety and effectiveness. The 510(k) should include the type, specifications and source of powder or other donning lubricant used on the gloves. Talc, cotton flock, and other non-absorbable materials are not acceptable as a lubricating, dusting or donning powder. Paragraph 4.3 of ASTM standards D 3578 or D 5250 requires the inside and outside surface of examination gloves to be free of talc. Lycopodium (club moss spores) and ground pine pollen are toxic and are not acceptable as powder on or in medical gloves.
Gloves should be subjected to leaching and washing or other appropriate reduction and removal processes for manufacturing material residue. Note that natural latex proteins are concomitant manufacturing materials as defined in 21 CFR §820.3(p) and must be controlled per §820.70(h).
Biocompatibility data should be submitted for all medical gloves. Because examination gloves are in direct contact with skin, a Primary Skin Irritation study and a Dermal Sensitization Study are appropriate. [See FDA CDRH ODE Blue Book G95-1, ISO TC 10993, and Chapter 5 for further guidance.] Biocompatibility tests should be performed on finished gloves. The data should be fully identified and presented in tables when feasible.
When a study such as a biocompatibility study is conducted by an internal or contract laboratory to establish a specification and/or obtain data for a submission to FDA, the device manufacturer should keep the original record of the results of the study on file at their factory or other readily accessible location. This original document should also include the name and address of the laboratory and device manufacturer; the device identity; and dates of testing.
If a change is made to gloves that could significantly affect safety or
effectiveness, such as adding or deleting powder; adding color, fragrance or a
claim to the labeling; or modifying an important process, a new and
complete 510(k) should be submitted. A new 510(k) usually is
not required if a manufacturer only does more of an existing process such as
extra leaching or washing and makes no claim or mention of this change on the
product labeling. A 510(k) submission for a new glove or for a modification to
an existing glove may also be submitted according to the guidance titled,
"The New 510(k) Paradigm - Alternative Approaches to Demonstrating
Substantial Equivalence in Premarket Notifications," available on the World
Wide Web at:
http://www.fda.gov/cdrh/ode/parad510.pdf
The submitter of a 510(k) for a modified glove should reference the original 510(k) number. Changes to patient examination gloves, labeling, packaging, processes, etc., are to be made according to the Quality System Regulation at 21 CFR §§820.40 and 820.70(b). FDA is proposing that patient examination gloves be reclassified as Class II devices. If patient examination gloves become Class II, changes to them must also meet the design control requirements of §820.30. [Changes made to documents under §820.30 automatically meet the requirements in §820.40.]
FDA relies on the voluntary standards issued by the American Society for Testing and Materials (ASTM) D 3578, D 3772 (finger cots) and D 5250 in assessing the parameters of patient examination gloves. ASTM D 5712 covers the Standard Test Method for the Analysis of Protein in Natural Rubber and Its Products. (Please see Chapter 12 on Voluntary Standards.)
The combination of microorganisms, starch, and moisture on examination gloves may result in microbial growth sufficient to cause discoloration, unpleasant odor and, occasionally, dangerous healthcare situations. (Bioburden and moisture control are primarily Quality System topics. However, because of the proposed requirement for expiration dating in §801.440(d) and problems with contaminated examination gloves, this control information is also printed here.)
To keep bioburden levels low on gloves:
It is obvious that moist, contaminated gloves cannot meet a significant expiration period or shelf life. Maintaining a long expiration period may require establishing a specification for moisture and bioburden; controlling bioburden; monitoring the moisture content of finished gloves; and ensuring that dispenser boxes be shrink-wrapped with plastic or otherwise be protected from moisture, and other contaminants. Changes in packaging and sealing should be evaluated, validated, and, in general, meet the change control requirements of the QS regulation.
Manufacturers that want to perform tests for bioburden may refer to Association for Advancement of Medical Instrumentation (AAMI) guidelines (USA FAX 703-276-0793) or to IES-RP-CC-005-87-T for Cleanroom Gloves and Finger Cots or consult a microbiological test laboratory.
Gloves intended to be sterilized should be controlled as noted above in order to keep their bioburden level well below the level that can be killed by the intended sterilization process.
Information on sterilization is located in Chapter 10 under Sterilization Notes. Finished sterile examination gloves should meet the ASTM standards for examination gloves, ASTM D 3578, D 3772, D 5250 or an equivalent standard, as appropriate. The manufacturer should have data demonstrating that the finished sterile examination gloves meet all specifications, including the AQL for pinholes. On a design qualifying basis, the sterilized gloves should pass the manufacturers protein, tensile strength, elongation, thickness, barrier, integrity, pinhole or leak acceptance test AQL, etc., after undergoing real time aging or accelerated aging such as for 7 days at 70 degrees Centigrade as described in ASTM D 3578, ASTM D 5250 or an equivalent standard, as appropriate.
If a manufacturer distributes examination gloves that are intended to be included in medical device kits, where the kit is to be sterilized, then the glove manufacturer should ensure that the gloves are capable of meeting the appropriate ASTM or equivalent glove standard after sterilization. Kit manufacturers and assemblers should be certain that gloves in their kits are cleared for marketing and can meet the appropriate FDA and ASTM or equivalent standards after sterilization by the method being used. Natural rubber latex gloves should be enclosed in their own packaging within the kit to avoid possible protein contamination of other devices. The kit must be labeled, as appropriate, per §801.437 for any latex in the devices or packaging; and eventually labeled per the final version of the proposed §801.440 regulation for the powder and protein on gloves and the expiration date (also see chapter 6).
PREMARKET NOTIFICATION SUBMISSION FORMAT
A suggested format for the submission of a premarket notification [510(k)] for patient examination gloves is presented on the next several pages. It is not a required format; however, it may be used as a guide for submitting the necessary information to FDA. This format will also increase the completeness and accuracy of your submission, and may reduce the time required to clear your gloves for marketing. Guidance for submitting the 510(k) information in this format starts on the next page.
Each 510(k) submission must be for only one type of glove such as a powder-free latex examination glove. Do not mix data for multiple types of gloves in one 510(k) submission. A 510(k) submission must be complete; that is, include all of the required information in your submission — do not state that the needed information is in another submission.
If examination gloves become Class II as proposed, than a 510(k) submission
for a new examination glove or for a modification to an existing examination
glove may be submitted according to the guidance titled, "The New 510(k)
Paradigm - Alternative Approaches to Demonstrating Substantial Equivalence in
Premarket Notifications," available on the World Wide Web at:
http://www.fda.gov/cdrh/ode/parad510.pdf
The applicant may continue to use the following format for applicant information, product identification, and data submissions associated with the new or modified glove. Be sure to also include the risk analysis, declaration of conformity, etc., as required by the paradigm.
Sample Format for a Premarket Notification [510(K)] for Examination Gloves
Please number the pages in your submission and attachments and include a table of contents. Do NOT include extraneous information such as copies of standards and details of test equipment. Please identify all attachments with the topic and the applicants name, street address, phone and FAX numbers.
1.0 Premarket Notification 510(k) Submission Applicant:
Name ______________________________________________________________________
Street Address _______________________________________________________________
Country ____________________________________________________________________
Phone No. _________________________________ FAX No. ________________________
| __________________________________ [Registration Number, from Form 2891(a)] |
A_________________________________ |
(Registration is required for all manufacturers,
importers, and repackers.
Listing is required by
U.S. manufacturers and by foreign manufacturers.
If the applicant
has submitted a registration form but has not received a registration number,
enter "applied for" in the registration blank above.)
1.1 Check the activity of the applicant:
|
[ ] Manufacturer |
[ ] Repacker |
[ ] Importer |
[ ] Consultant |
[ ] Other |
Describe Other:
_______________________________________________________________
____________________________________________________________________________
____________________________________________________________________________
1.2 The applicant must include the name of the current manufacturer under 5.0 below. The 510(k) is a permanent record, and the name will not be changed or transferred by FDA.
1.3 Manufacturers that have a contact person within the firm as well as a contact (consultant, importer, etc.) in other locations should give the names of both persons below.
Contact Person in Firm:__________________________________________________________
Phone No. ____________________________ FAX No. _________________________________
Other Contact Person:___________________________________________________________
Phone No. _____________________________ FAX No.________________________________
2.0 Truthful and Accurate Statement: As shown below, include a statement identifying your capacity or position in the company and the manufacturer’s name certifying that all information submitted in the 510(k) is truthful and accurate and that no material fact has been omitted.
Sample
PREMARKET NOTIFICATION
TRUTHFUL AND ACCURATE STATEMENT
[As required by 21 CFR 807.87(j)]
| I certify that, in my capacity as |
(________________________________________________) |
of |
|
I believe to the best of my knowledge, that all data and information submitted in the premarket notification are truthful and accurate and that no material fact has been omitted.
|
|
|
|
|
|
|
|
The statement must be signed by a responsible person of the company required to submit the premarket notification — not a consultant for the submitter.
* For a new submission, do not fill in the 510(k) number. The Food and Drug Administration will fill in this blank with your 510(k) number when the number is assigned.
3.0 Indications for Use Statement: Include the following or equivalent Indications for Use page. The information, data and labeling claims in the entire 510(k) submission must support and agree with the Indications for Use statement.
INDICATIONS FOR USE
Applicant:___________________________________________________________________
510(k) Number (if known): * ____________________________________________________
Device Name: ________________________________________________________________
Indications For Use:
* For a new submission, do NOT fill in the 510(k) number.
4.0 Glove Proprietary or Trade Name:
___________________________________________
4.1 Modification: If this submission is for a modification of an examination glove cleared by FDA for marketing, include the 510(k) number of the cleared glove: ___________________________
5.0 Name and Location of ACTUAL Manufacturer:
Name ______________________________________________________________________
Address ____________________________________________________________________
Country ____________________________________________________________________
Phone No. _____________________________ FAX No. _________________________
___________________________________ [Registration Number, from Form 2891(a)] |
A__________________________________ (Device Listing Number, from Form 2892) |
6.0 Labels, Labeling, and Advertising: The labeling must include basic information (See Chapter 6 for guidance); and labeling must include the appropriate caution statements and an expiration date as included in proposed §801.440. Include identified copies of all labeling or proposed labeling, including promotional literature. The labeling should contain the common, generic or scientific name of the polymer of which the glove is composed. "Synthetic" used alone has no meaning. If you make any specific claims for your gloves, include data to substantiate the claims in this format or in identified attachments. Puffery, ambiguous, or unsubstantiated claims such as extra thick, low protein, or super sensitive are not allowed. Labeling, labeling claims and data must be consistent with the Indications for Use statement.
7.0 Classification Information
7.1 Device Class: I [Proposed Class II ]
| 7.2 Substantial Equivalent Device Description: | ( check one ) |
| [ ] Patient Examination Glove, powdered | 21 CFR 880.6250 proposed |
| [ ] Patient Examination Glove, powder-free | 21 CFR 880.6251 proposed |
7.3 Product Code: |
( check one ) |
| [ ] Vinyl - 80LYZ | [ ] Latex - 80LYY |
| [ ] Synthetic Polymer - 80LZA | [ ] Nitrile - 80LZA |
| [ ] Specialty - 80LZC | [ ] Finger Cot - 80LZB |
| [ ] Other - 80FMC | |
| If Finger Cot or "Other," identify
material:
______________________________________ ________________________________________________________________________ | |
8.0 Specifications (of your specific LATEX or Synthetic "xxxx" Polymer Glove (except vinyl):
| Overall Length: | ________mm minimum | |
| Width: | ________mm minimum (for medium glove) | |
| Palm Thickness: |
________mm minimum |
Finger Thickness: ________mm
minimum after aging |
| Tensile Strength: | _____________ Mpa minimum | _______________Mpa minimum |
| Ultimate Elongation: | ______________% minimum | ______________ % minimum |
| Your Pinhole AQL | ______________ | ______________@70°C for 7 days* |
* or equivalent aging to show integrity when used.
Does the above data for your examination gloves meet ALL the current specifications listed under the ASTM Specification D 3577.?
YES ___ NO ___ If NO, explain why in an identified attachment.
IF VINYL: Your Pinhole AQL before ________
and after aging ________.
Do the vinyl examination gloves meet
ALL the current specifications listed under ASTM Specification
D 5250? YES ___ NO ___ If NO, explain why in
an identified attachment.
8.1 Specialty, Chemotherapy Gloves:
For chemotherapy or other specialty gloves, include in an identified attachment any additional specifications needed to support your labeling claims.
9.0 Quality Assurance Testing (of Finished Gloves):
Finished product quality assurance testing for physical properties such as tensile strength and elongation; dimensions such as length, width, and thickness; chemical tests such as pH; moisture; powder residues; and leak testing are important for assuring a quality product. Visual tests such as color, material uniformity, etc., are also commonly performed. ASTM D 3578, standard for latex examination gloves, and D 5250, standard for vinyl examination gloves, refer to test methods and sampling procedures. For production barrier, integrity, pinhole or leak testing, the sampling and testing should conform to the test methods and AQL established by the manufacturer under their quality system acceptance criteria in 21 CFR 820.181.
Does your quality assurance test results for the examination gloves conform to ALL ASTM D 3578, or D 5250 procedures?
YES ____ NO ____
Describe your quality assurance procedures in an identified attachment. The attachment should describe the test methods and acceptance criteria such as sampling procedures, and acceptance quality levels (AQL). Reference any standard test methods that are used.
9.1 Specialty, Chemotherapy Gloves Data:
In addition to the data in 9.0, include data in an identified attachment to show that the gloves are safe and effective for handling chemotherapy agents specified in the labeling or any other special claim.
10.0 Sterility: Are these examination gloves labeled as sterile? YES _____ NO _____
If YES, state sterilization method (radiation, gas, etc.) used: _________________________
10.1 Sterility Assurance Level (SAL): _________________________________________
(The SAL is the statistical probability of a glove not being sterile after going through the validated sterilization cycle. The SAL must be 10-6 or better for a sterile glove.)
10.2 How was the sterilization cycle validated?
__________________________________________________________________________
__________________________________________________________________________
__________________________________________________________________________
__________________________________________________________________________
__________________________________________________________________________
10.3 If Radiation sterilization, dose in Kilograys ____________________
10.4 If EtO Sterilization, level of residue in parts per million (PPM) for:
Ethylene Oxide ________________________________
Ethylene Chlorohydrin __________________________
Ethylene Glycol _______________________________
10.5 Describe packaging used to maintain sterility: _______________________________
__________________________________________________________________________
__________________________________________________________________________
10.6 Sterilizer Name _________________________________________________________
Address ___________________________________________________________________
Address ___________________________________________________________________
Country ____________________________________________________________________
Phone No. _____________________________ FAX No. _____________________________
Registration Number [from Form 2891(a)] _____________________
If sterilization is done by a contractor, the glove manufacturer must have a contract with the contract sterilizer that meets the requirements of §801.150(e). An importer may need two written agreements: one with the foreign manufacturer; and a second agreement with the contract sterilizer.
11.0 FORMER Release Powder or Chemical: (If none is used, state none and skip to 12.)
Release Powder or Chemical __________________________________________________
Supplier ___________________________________________________________________
Specifications _______________________________________________________________
12.0 Dusting or Donning Powder:
(Skip to 13 if "powder-free")
(ASTM standards do
not allow Talc on the surface of medical gloves.)
U.S.P. Absorbable Dusting Powder used? YES _____ NO _____.
If non-U.S.P. absorbable dusting powder is used, then state the:
Powder Type ________________________________________________________________
Supplier ____________________________________________________________________
Brand Name _________________________________________________________________
Specifications ________________________________________________________________
____________________________________________________________________________
12.1 Weight of Donning Powder:
Weight of all types of powder on finished powdered glove _______ +/- _______ milligrams per glove. FDA is recommending that donning powder not exceed 120 mg per glove. Powder should be measured by ASTM D-6124, June 24, 1999.
13.0 Weight of Powder-free Residue:
Weight of all types of residual or trace powder on finished powder-free glove _____ +/- _____ mg per glove determined by ASTM D 6124. Residue should not exceed 2 mg per glove or the limit in the ASTM standard.
If the gloves are "powder-free," and the process includes any mold / former release or donning powder, then the applicant should provide items 13.1 through 13.5 below.
If the gloves are "powder-free" and the process does NOT include any powder, then the applicant should complete items 13.4 and 13.5 below.
13.1 Describe the powder(s) introduced at any stage of the glove manufacturing process.
__________________________________________________________________________
__________________________________________________________________________
13.2 In an identified attachment, describe in detail the process to remove the added powder(s).
13.3 In an identified attachment, include and describe the finished glove release specification supporting the "powder-free" claim and a brief summary of final product testing to ensure finished gloves meet this specification. (You should use the ASTM D 6124 method or an equivalent standard for measuring residual or trace powder.)
13.4 Completely describe in an identified attachment how the glove is designed or manufactured to compensate for the lack of donning powder, or reasons why compensation is not necessary, including a full characterization (e.g., chemical identity, specifications, biocompatibility) of any material such as silicone or polymer coating on the glove to facilitate glove donning.
If a donning lubricant is used, state the exact composition and include biocompatibility data for the lubricant in an identified attachment; also state the name, manufacturer, and address below:
Lubricant Generic Name ________________________________________________________
Lubricant Brand Name(s) _______________________________________________________
Lubricant Manufacturer _________________________________________________________
Address _____________________________________________________________________
Phone No. ___________________________ FAX No. _____________________________
13.5 You should certify that your finished "powder-free" gloves meet ASTM D 3578 standard or equivalent for natural rubber latex or ASTM D 5250 standard or an equivalent standard for vinyl. On a design qualifying basis, the gloves should meet the manufacturers barrier, integrity, pinhole or water leak test and acceptance criteria after being subjected to real time aging or to the ASTM accelerated aging test of 7 days at 70°C. (You may refer to data in 8, 9 and 10 above.)
14.0 Protein Level of Natural Rubber Latex Gloves:
Water soluble protein measured by ASTM D 5712 yielded _________ +/- ________ micrograms per glove. FDA is recommending that protein should not exceed 1200 micrograms on any size glove. The sensitivity of ASTM Lowry test method does not support claims below 300 µg per glove (derived from 50µg/gm of glove sensitivity X 6 grams for a typical glove = 300).
14.1 ASTM D 5712-95 Standard Test Method for the Analysis of Protein in Natural Rubber and Its Products was used to determine the protein level? YES _____ NO _____
If NO, include a complete description of the test method used and data showing how it correlates with the ASTM method.
14.2 The protein testing was performed on the final finished gloves that have undergone real time aging or accelerated aging per ASTM D 3578: YES _____ NO _____
14.3 Include the sampling method and sample size.
14.4 Include your acceptance / rejection criteria.
14.5 Include a summary of test results from samples of at least one lot of gloves using ASTM D 5712-95, that supports your stated protein level.
14.6 Include the chemical identity, biocompatibility, and specification for ANY material added to and remaining on the glove to reduce total water extractable proteins. (You may refer to 18 Biocompatibility below.)
In an identified attachment, describe the manufacturing process steps that are used to achieve the protein level.
15.1 In an identified attachment, include a summary of quality control procedures that contains the following 15.2 to 15.6:
15.2 The specification or set point for the glove protein content that will be used for quality control during routine production:
15.3 Specify if a test method other than ASTM D 5712-95 will be used for determining protein content during routine production: YES _____ NO _____;
15.4 If 15.3 is YES, include data correlating the routine quality control method to the ASTM D 5712-95 method; and
15.5 Specify the frequency the ASTM D 5712-95 method will be used to verify performance of the routine method. _____________________________________________________________
__________________________________________________________________________
16.0 Chemical Sensitivity Claim: Include your chemical sensitivity claim, if any, in an identified attachment with supporting data. For guidance, please refer to the document titled, "Draft Guidance on the Content and Format of Premarket Notification [510(k)] Submissions for Testing for Skin Sensitization to Chemicals in Latex Products," available on the World Wide Web at: http://www.fda.gov/cdrh/ode/944.html.
17.0 Color or Flavor Additives:
Any color additive or flavor additive used in manufacturing medical gloves must be identified. Provide the chemical name and composition of the color or flavor additive used. Include in an identified attachment in step 18 biocompatibility data to support safe use of the additive.
Biocompatibility data should be submitted for examination gloves. Perform biocompatibility tests on finished gloves and include the result in an identified attachment. Use tables where feasible. Because examination gloves contact skin, skin irritation and dermal sensitization tests are considered appropriate.
19.0 Expiration Date or Quality at Delivery:
FDA is proposing that labeling contain an expiration date and, if the proposed regulation becomes final, an expiration date and data to support it will be required in 510(k) submissions. After the proposed regulation becomes a final rule, respond to 19.1; in the interim you should respond to 19.1 or 19.2.
19.1 Expiration Date. For the gloves covered by this 510(k) submission, include the length of the expiration period in months and years in your label claim for which you have valid data to support. [See chapter 6 and proposed §801.440(d) for guidance. Data must be maintained by the manufacturers to support an optional or required (proposed) expiration date of their gloves.]
19.2 Quality at Delivery. If you do not complete 19.1 above, submit data to show that your gloves meet the applicable ASTM or equivalent standard requirements including pinhole requirements after real time aging for at least three months or after accelerated aging for 7 days at 70 degrees centigrade. (No label claim or expiration date is involved or allowed for this minimal data.)
20.0 Other Claims Requiring Data:
List any other claim that needs data to support.______________________________________
___________________________________________________________________________
___________________________________________________________________________
20.1 List in a table format the appropriate assay and timeframe for the evaluation that you used for each of the claims in 20.0.
21.0 510(k) Summary/Statement Requirement:
(See Chapter 7, 510(k) Summary and Statement Information.)
You MUST include on a SEPARATE sheet(s) your name, address and either:
You may not begin commercial distribution of a device in the United States until you receive a letter from FDA stating that your medical glove was found to be substantially equivalent. Marketing the device prior to a finding of substantial equivalence would render the device adulterated under §501(f)(1)(B) of the FD&C Act and would be subject to enforcement action by the FDA.
DEFINITION AND
REQUIREMENTS
VOLUNTARY STANDARDS
STERILITY,
BIOBURDEN AND MOISTURE
PREMARKET
NOTIFICATION [510(k)] SUBMISSION FORMAT *
New 510(k) Paradigm
Sample Format for a Premarket Notification [510(K)] for Surgeon’s Gloves
Premarket Notification 510(k) Submission Applicant
Sample Premarket Notification Truthful and Accurate Statement
Indications for Use Statement
Glove Proprietary or Trade Name
Name and Location of ACTUAL Manufacturer
Labels, Labeling, and Advertising
Classification Information
Specifications
Quality Assurance Testing
Sterility
Former Release Powder or Chemical
Absorbable Dusting or Donning Powder
Weight of Powder-free Residue
Protein Level of Latex Gloves
Protein Control
Chemical Sensitivity Claim
Color or Flavor Additives
Glove Biocompatibility
Expiration Date or Quality at Delivery
Other Claims Requiring Data
510(k) Summary/Statement Requirement
Under the proposed rule, surgeon's gloves would be Class II (special controls) devices and would be identified as follows:
Surgeon’s Gloves, powdered (proposed §878.4460). A powdered surgeon’s glove is a disposable device made of natural rubber latex or synthetic material that bears powder to facilitate donning, and it is intended to be worn on the hands, usually in surgical settings, to provide a barrier against potentially infectious materials and other contaminants.
The Class II special controls are:
User labeling requirements in 21 CFR §801.440.
Surgeon’s Gloves, powder-free (proposed § 878.4461). A powder-free surgeon’s glove is a disposable device made of natural rubber latex or synthetic material that may bear a trace amount of glove powder and is intended to be worn on the hands, usually in surgical settings to provide a barrier against potentially infectious materials and other contaminants.
The Class II special controls are:
Various types of surgeon’s gloves are described in Chapter 3, Product Identification. The classification panel number and product code for all surgeon’s gloves is 79KGO. Surgeon’s gloves are subject to the registration, listing, labeling, premarket notification [(510(k)], medical device reporting, QS regulation and other requirements of the FD&C Act. A 510(k) clearance letter is needed from FDA before a medical glove may be distributed in the United States (U.S.).
Surgeon’s gloves should be sterile when offered for sale to end users such as hospitals, clinics, surgeon’s, etc. FDA will not accept a 510(k) submission for a non-sterile surgeon’s glove.
Powder-free surgeon’s gloves may be lubricated with small amounts of silicone or other suitable lubricant or be coated with a non-tacky polymer. A submission should include a full characterization of the lubricant or coating such as the chemical identity, specifications and biocompatibility.
Absorbable donning or dusting powder from powder manufacturers that have obtained an approved new drug application (NDA), abbreviated new drug application (ANDA), or premarket approval application (PMA) must be used on powdered surgeon’s gloves. A list of companies that have obtained either an NDA or a PMA approval for U.S.P. absorbable donning powder for use on surgeon’s gloves is in Chapter 4, Glove Lubricants. Note that paragraph 5.3 of ASTM standard D 3577 requires that the inside and outside surface of surgical gloves be free of talc.
Surgeon’s gloves should be shown to be biocompatible. Manufacturers should subject gloves to leaching and washing or other appropriate reduction and removal processes for manufacturing material residues per the Quality System requirements in 21 CFR 820.3(p) and 820.70(h). Also note that latex proteins are concomitant manufacturing materials as defined in §820.3(p) and must be controlled per §820.70(h). (Latex gloves may also be coated with certain synthetic polymers to reduce the availability of latex proteins and processing chemicals and reduce the need for donning lubricants.)
All surgeon’s gloves should be subjected to a primary skin irritation study and a dermal sensitization study. See FDA, CDRH, ODE Blue Book G95-1, ISO TC 10993 and chapter 5 for further guidance. Biocompatibility tests should be performed on finished sterile gloves. The data should be fully identified and presented in tables when feasible. When a study such as a biocompatibility study is conducted by an internal or contract laboratory to establish a specification and/or obtain data for a submission to FDA, the device manufacturer should keep the original record of the results of the study on file. This original record must also include the name and address of the laboratory and device manufacturer, the device identity, and dates of testing. These verification records are part of the QS design history file required by §820.30(j).
If a new labeling claim is added, or a change is made, to gloves that could
significantly affect safety or effectiveness, such as adding or deleting powder,
adding color, or modifying an important process, a new, complete 510(k) should
be submitted. A new 510(k) usually is not required if a manufacturer only does
more of an existing process such as extra leaching or washing and makes no
special claim or mention of this change on the product labeling. A 510(k)
submission for a new glove or for a modification to an existing glove may be
submitted according to the guidance titled, "The New 510(k) Paradigm -
Alternative Approaches to Demonstrating Substantial Equivalence in Premarket
Notifications," available on the World Wide Web at:
http://www.fda.gov/cdrh/ode/parad510.pdf
The submitter of a new 510(k) for a modified glove should reference the 510(k) number for the original glove. For surgeon's gloves, changes to materials, gloves, processing, labeling, testing, packaging, etc., must be made according to QS design change control requirements in §§820.30, 820.40 and 820.70(b). [Changes made to documents under §820.30 automatically meet the requirements in §820.40.]
FDA relies on the voluntary standard D 3577 issued by the American Society for Testing and Materials (ASTM) in assessing the parameters of surgical gloves. Please see Chapter 12, Voluntary Standards.
The method that FDA uses for their pinhole leak testing, along with sampling plans, was published at 21 CFR §800.20 (see chapter 11). Glove manufacturers need to establish their test methods for barrier, integrity, leak or pinhole testing as required by Quality System §820.181 (acceptance criteria) and also as required by §820.30.
STERILITY, BIOBURDEN AND MOISTURE
Gloves intended to be sterilized should be controlled in order to keep their bioburden level well below the level that can be killed by the intended sterilization process. Information on sterilization is located in Chapter 10 under Sterilization Notes.
The combination of microorganisms, starch, and moisture on medical gloves may result in microbial growth sufficient to cause discoloration, an unpleasant odor and, occasionally, dangerous endotoxins.
To keep bioburden levels low on gloves:
Finished sterile gloves should meet the ASTM standard for surgeon’s gloves, ASTM D 3577 or an equivalent standard, as appropriate. The manufacturer should have data demonstrating that the finished sterile surgeon’s gloves pass their elongation, tensile and barrier, integrity, leak or pinhole test and acceptance criteria as required by 21 CFR §820.181. (FDA uses a 1000 milliliter water leak test in accordance with the sample plan and test method in 21 CFR §800.20. On a design qualifying basis to show that they will meet the intended use or user/patient needs, the sterilized gloves should meet the manufacturers acceptance criteria for all parameters including barrier integrity after undergoing real time aging for a suitable period or suitable accelerated aging such as for 7 days at 70 degrees Centigrade as described in ASTM D 3577, or an equivalent standard. (The accelerated aging conditions are expected to change when ongoing studies are completed.) (Routine leak testing during production should be done on non-aged gloves.)
PREMARKET NOTIFICATION [510(k)] SUBMISSION FORMAT
A suggested format for the submission of a premarket notification [510(k)] for surgeon’s gloves is presented on the next several pages. It is not a required format; however, it may be used as a guide for submitting the required information to FDA. This format should increase the completeness and accuracy of your submission and reduce the time required to clear your gloves for marketing.
Each 510(k) submission must be for only one type of glove such as a powder-free latex surgeon’s glove. Do not mix data for multiple types of surgeon’s gloves in one 510(k) submission. A 510(k) submission must be complete; that is, include all of the required information in your submission — do not state that the needed information is in another submission.
A 510(k) submission for a new surgeon’s glove or for a modification to an existing surgeon‘s glove may be submitted according to the guidance titled, "The New 510(k) Paradigm - Alternative Approaches to Demonstrating Substantial Equivalence in Premarket Notifications," available on the World Wide Web at: http://www.fda.gov/cdrh/ode/parad510.pdf
The applicant may continue to use the following format for applicant information, product identification, and data submissions associated with the new or modified glove. Be sure to also include the risk analysis, declaration of conformity, etc., as required by the paradigm.
Sample Format for a Premarket Notification [510(K)] for Surgeon’s Gloves
Please number the pages in your submission and attachments and include a table of contents. Do NOT include extraneous information such as copies of standards and details of test equipment. Please identify all attachments with the topic and the applicants name, street address, phone and FAX numbers.
1.0 Premarket Notification 510(k) Submission Applicant:
Name ______________________________________________________________________
Street Address _______________________________________________________________
Country _____________________________________________________________________
Phone No. ___________________________ FAX No. _____________________________
___________________________________ [Registration Number, from Form 2891(a)] |
A__________________________________ (Device Listing Number, from Form 2892) |
(Registration is required for all manufacturers, importers, and
repackers.
Listing is required by U.S. manufacturers and by foreign
manufacturers.
If the applicant has submitted a registration form but has not
received a registration number, enter "applied for" in the registration
blank.)
1.1 Check the activity of the applicant:
| [ ] Manufacturer | [ ] Repacker | [ ] Importer | [ ] Consultant | [ ] Other |
Describe Other: _____________________________________________________________
__________________________________________________________________________
1.2 Applicant must include the name of the current manufacturer under 5.0 below. The 510(k) is a permanent record, and the name in block #1.0 will not be changed or transferred by FDA.
1.3 Manufacturers that have a contact person within the firm as well as a contact such as a consultant, importer, etc., in other locations should give the names of both persons below.
Contact Person in Firm: _______________________________________________________
Phone No. _______________________ FAX No. ___________________________________
Other Contact Person: _________________________________________________________
Phone No. _______________________ FAX No. ___________________________________
2.0 Truthful and Accurate Statement: As shown below, include a statement identifying your capacity or position in the company and the manufacturer name certifying that all information submitted in the 510(k) is truthful and accurate and that no material fact has been omitted.
Sample
PREMARKET NOTIFICATION
TRUTHFUL
AND ACCURATE STATEMENT
[As required by 21 CFR 807.87(j)]
| I certify that, in my capacity as |
(________________________________________________) |
| of |
(
__________________________________________________________________), |
I believe to the best of my knowledge, that all data and information submitted in the premarket notification are truthful and accurate and that no material fact has been omitted.
|
______________________________________ |
|
______________________________________ |
|
______________________________________ |
|
______________________________________ |
The statement must be signed by a responsible person of the company required to submit the premarket notification — not a consultant for the submitter.
* For a new submission, do not fill in the 510(k) number. The Food and Drug Administration will fill in this blank with your 510(k) number when the number is assigned.
3.0 Indications for Use Statement: Include the following or equivalent Indications for Use page. (See chapter 7.) The information, data and labeling claims in the entire 510(k) submission must support and agree with the Indications for Use statement.
INDICATIONS FOR USE
Applicant: __________________________________________________________________
510(k) Number (if known):* ____________________________________________________
Device Name: _______________________________________________________________
Indications For Use:
* For a new submission, do NOT fill in the 510(k) number.
4.0 Glove Proprietary or Trade Name: __________________________________________
4.1 Modification: If this submission is for a modification of a surgeon’s glove cleared by FDA for marketing, include the 510(k) number of the cleared glove: _____________________________
5.0 Name and Location of ACTUAL Manufacturer:
Name ______________________________________________________________________
Address ____________________________________________________________________
Country ____________________________________________________________________
Phone No. ____________________________ FAX No. ______________________________
___________________________________ [Registration Number, from Form 2891(a)] |
A__________________________________ (Device Listing Number, from Form 2892) |
6.0 Labels, Labeling, and Advertising: The labeling must include basic information (See Chapter 6 for guidance) and labeling must include the appropriate caution statements and a expiration date as included in proposed §801.440 when it becomes final. Include identified copies of all labeling or proposed labeling, including promotional literature. The labeling should contain the common, generic or scientific name of the polymer of which the glove is composed. "Synthetic" used alone has no meaning. If powdered, labeling should include the statement, "Powdered with Absorbable Dusting Powder." The labeling on the envelope or package containing the surgeon’s gloves must include a caution statement directing the user to remove the lubricating powder after donning the gloves (see chapter 6 or 36 FR p.9475, May 25, 1971, reprinted at end of chapter 4).
If you make any specific claims for your gloves, include data to substantiate the claims in this format or in identified attachments. Ambiguous or unsubstantiated claims such as low protein, super strong, extra thick, super sensitive, micro thin, etc., are not allowed.
Labeling, labeling claims and data must be consistent with the "Indications for Use" statement.
7.0 Classification Information:
7.1 Device Class: I [Proposed for Class II]
| 7.2 Substantial Equivalent Device Description: | ( check one ) |
| [ ] Surgeon’s Glove, powdered | 21 CFR §878.4460 proposed |
| [ ] Surgeon’s Glove, powder-free | 21 CFR §878.4461 proposed |
7.3 Type: |
( check one ) |
| [ ] Type 1 - gloves compounded primarily from natural rubber latex, or |
| [ ] Type 2 - gloves compounded primarily from rubber cement or synthetic rubber latex. |
7.4 Product Code: |
( check one ) |
[ ] Surgeon’s Glove 79KGO
[ ] Autopsy Gloves 79LYU
[ ] Glove Liners
79KGO
[ ] Radiographic Protection 90IWP
[ ] Leak Detector 79LDQ
| 7.5 Composition of Gloves: | ( check one ) |
| [ ] Latex | [ ] Latex Polymer Coated |
| [ ] Synthetic Polymer (See 6.0 above) | [ ] Co-Polymer |
| [ ] Other | |
Describe other: ___________________________________________________________
________________________________________________________________________
| Overall Length: | ________ mm minimum | |
| Width: | ________ mm minimum (for medium glove) | |
| Palm Thickness: | ________ mm minimum | |
| Finger Thickness: | ________ mm minimum | |
Before Aging |
After Aging* @ 70°C for 7 days | |
| Tensile Strength: | _________ Mpa minimum | _______________ Mpa minimum |
| Ultimate Elongation: | _________ % minimum | _______________ % minimum |
| Your Pinhole AQL: | _________ | _______________ |
* Or equivalent aging to show barrier integrity when used.
8.1 IF LATEX GLOVES: Does the above data for your latex surgeon’s gloves meet ALL the current specifications listed in the ASTM Standard D 3577?
YES ____ NO ____ If NO, explain why in an identified attachment; and state the equivalent standard to which your gloves conform.
8.2 IF SYNTHETIC POLYMER GLOVES: Does the above data for your polymer surgeon’s gloves meet ALL the current specifications listed under ASTM Standard D 3577 or specific standard for gloves composed of the specific synthetic polymer?
YES ____ NO ____ If NO, state the glove composition and all of your specifications in an identified attachment; and state the equivalent standard to which your gloves conform.
9.0 Quality Assurance Testing (of Finished Gloves):
Finished product quality assurance testing for physical properties such as tensile strength and elongation; dimensions such as length, width, and thickness; chemical tests such as pH and heavy metals; moisture; powder residues; and leak testing are important for assuring a quality product. Visual tests such as color, material uniformity, etc., are also commonly performed. ASTM D 3577, the voluntary standard for latex surgeon’s gloves, refers to test methods and sampling procedures. For production barrier, integrity, pinhole or leak testing, the sampling and testing should conform to the test methods and AQL established by the manufacturer under their quality system acceptance criteria in §820.181.
Does your quality assurance result conform to all ASTM D
3577 procedures and the FDA water leak test requirements? YES ____ NO ____
Describe your quality assurance procedures in an identified attachment. The
attachment should describe the test methods and test criteria such as sampling
procedures, and acceptance quality levels (AQL). Reference any standard test
methods that are used.
9.1 Specialty Surgeon's Gloves:
If the submission contains a special claim, in addition to the data in 9.0, include data in an identified attachment to show that the specialty surgeon's gloves are safe and effective for the special claim.
State sterilization method (radiation, gas, etc.) used: ________________________________
10.1 Sterility Assurance Level (SAL): ___________
(The
SAL is the statistical probability of a glove not being sterile after going
through the validated sterilization cycle. The SAL must be 10-6 or
better for a sterile glove.)
10.2 How was the sterilization cycle validated? ___________________________________
___________________________________________________________________________
___________________________________________________________________________
___________________________________________________________________________
___________________________________________________________________________
___________________________________________________________________________
10.3 If Radiation sterilization, dose in Kilograys: ___________________________________
10.4 If EtO Sterilization, reference the methods for determining residues and state the level of residue in parts per million (PPM):
Test Methods ________________________________________________________________
Ethylene Oxide _______________________________________________________________
Ethylene Chlorohydrin _________________________________________________________
Ethylene Glycol ______________________________________________________________
10.5 Describe packaging used to maintain sterility: ________________________________
___________________________________________________________________________
___________________________________________________________________________
___________________________________________________________________________
___________________________________________________________________________
10.6 Sterilizer: Name _________________________________________________________
Address ____________________________________________________________________
Address ____________________________________________________________________
Country _____________________________________________________________________
Phone No. _____________________________ FAX No. ______________________________
Registration Number [from Form 2891(a)] __________________________________________
If the sterilization is done by a contractor, the glove manufacturer must have a contract with the contract sterilizer that meets the requirements of § 801.150(e). An importer may need two contracts: one with the foreign manufacturer; and a second agreement with the contract sterilizer.
11.0 FORMER Release Powder or Chemical: (If none is used, state none and go to 12.)
Release Powder or Chemical ___________________________________________________
Supplier ____________________________________________________________________
Specifications ________________________________________________________________
12.0 Absorbable Dusting or Donning
Powder: (Skip to 13 if "powder-free")
[ASTM standards do
not allow Talc on the surface of medical gloves.]
Supplier ____________________________________________________________________
Address ____________________________________________________________________
Brand Name _________________________________________________________________
NDA, ANDA or PMA number ___________________________________________________
Specifications ________________________________________________________________
12.1 For Finished Powdered Gloves:
Weight of all types of powder on a glove _______ +/- _______ milligrams per
glove.
FDA is recommending that the weight of powder not exceed 120
mg per glove.
13.0 Weight of Powder-free Residue:
Weight of all types of residual or trace powder on a glove _______ +/- _______ milligrams per glove determined by ASTM D 6124. The weight of trace powder should not exceed 2 mg.
If the gloves are powder-free, and the manufacturing process includes any mold / former release or donning powder, then the applicant must provide items 13.1 through 13.5 below.
If the gloves are powder-free and the manufacturing process does NOT include any powder, then the applicant should complete items 13.4 and 13.5 below.
13.1 Describe the powder(s) introduced at any stage of the glove manufacturing process:
___________________________________________________________________________
___________________________________________________________________________
___________________________________________________________________________
13.2 In an identified attachment, describe in detail the process to remove the added powder(s).
13.3 In an identified attachment, include and describe the finished glove release specification supporting the "powder-free" claim and a brief summary of final product testing to ensure finished gloves meet this specification. (Manufacturers should use the ASTM D 6124 or an equivalent method for measuring residual or trace powder.)
13.4 Completely describe in an identified attachment how the glove is designed or manufactured to compensate for the lack of donning powder, or reasons why the compensation for lack of donning powder is not necessary, including a full characterization such as chemical identity, specifications, biocompatibility of any material such as silicone or polymer coating on the glove to facilitate glove donning.
If a donning lubricant is used, state the exact composition and include biocompatibility data for the lubricant in an identified attachment; and state the name, manufacturer, and address below:
Lubricant Generic Name _______________________________________________________
Lubricant Brand Name(s) ______________________________________________________
Lubricant Manufacturer ________________________________________________________
Address ____________________________________________________________________
Address ____________________________________________________________________
Phone No. ________________________________ FAX No. __________________________
13.5 You should certify that your finished sterile "powder-free" gloves meet ASTM D 3577 standard or an equivalent standard for latex and polymers. On a design qualifying basis, the gloves should meet the manufacturers barrier, integrity, pinhole or water leak test and acceptance criteria after being subjected to real time aging or to the ASTM accelerated aging test of 7 days at 70°C. (You may refer to data in 8, 9 and 10 above.)
FOR SYNTHETIC POLYMER GLOVES, SKIP TO STEP 16.
14.0 Protein Level of Latex Gloves:
Water-soluble protein measured by ASTM D 5712 yielded ________ +/- _______ micrograms per glove. FDA is recommending that the protein not exceed 1200 µg on any size glove. The sensitivity of ASTM Lowry test method does not support claims below 300 µg per glove (derived from 50µg/gm of glove sensitivity X 6 grams for a typical glove = 300).
14.1 ASTM D 5712-95 Standard Test Method for the Analysis of Protein in Natural Rubber and Its Products was used to determine the protein level. YES _____ NO _____
If NO, include a complete description of the test method used and data showing how it correlates with ASTM D 5712.
14.2 The protein testing was done on the final finished gloves that have undergone real time aging or accelerated aging per ASTM D 3577: YES _____ NO _____
14.3 In an identified attachment, include the sampling method and sample size.
14.4 In an identified attachment, include the acceptance/rejection criteria.
14.5 Include in an identified attachment a summary of test results from samples of at least one lot of gloves using ASTM D 5712-95, that supports your stated protein level.
14.6 Include the chemical identity, biocompatibility, and specification for ANY material added to and remaining on the glove to reduce total water extractable proteins. (You may refer to 18 Biocompatibility below.)
15.0 Protein Control: In an identified attachment, describe the manufacturing process steps that are used to achieve the claimed protein level.
15.1 In an identified attachment, include a summary of quality control procedures that contains the following 15.2 to 15.5:
15.2 The specification or set point, for the glove protein content that will be used for quality control during routine production;
15.3 Specify if a test method other than ASTM D 5712-95 will be used for determining protein content during routine production: YES _____ NO _____;
15.4 If 15.3 is YES, include data correlating the routine quality control method to the ASTM D 5712-95 method; and
15.5 Specify the frequency the ASTM D 5712-95 method will be used to verify performance of the routine method. _______________________________________________________________
16.0 Chemical Sensitivity Claim:
Include your chemical sensitivity claim, if any, in an identified attachment
with supporting data.
For guidance, please refer to the document titled,
"Draft Guidance on the Content and Format of Premarket Notification [510(k)]
Submissions for Testing for Skin Sensitization to Chemicals in Latex
Products," available on the World Wide Web at: http://www.fda.gov/cdrh/ode/944.html.
17.0 Color or Flavor Additives:
Any color additive or flavor additive used in manufacturing medical gloves must be identified. Provide the chemical name and composition of the color or flavor additive used. Include in an identified attachment in step 18 biocompatibility data to support safe use of the additive.
Perform biocompatibility tests on finished sterile gloves and include the results in an identified attachment. Use tables where feasible for data. The applicant should cite the specific test methods used and state the results obtained, i.e., "...under conditions of the tests, the finished sterile gloves were not (or were ) sensitizing and were not (or were ) irritating." (See chapter 5.)
19.0 Expiration Date or Quality at Delivery:
FDA is proposing that labeling contain an expiration date and, if the proposed regulation becomes final, an expiration date and data to support it will be required in 510(k) submissions. After the proposed regulation becomes a final rule, respond to 19.1; in the interim you should respond to 19.1 or 19.2 to help show your gloves meet the intended use and user/patient needs.
19.1 Expiration Date. For the gloves covered by this 510(k) submission, include the length of the expiration period in months and years in your label claim for which you have valid data to support. [See chapter 6 and proposed §801.440(d) for guidance. Data must be maintained by the manufacturers to support an optional or required (proposed) expiration date of their gloves.]
19.2 Quality at Delivery. If you do not complete 19.1 above, submit data to show that your gloves meet the applicable ASTM or equivalent standard requirements including pinhole requirements after real time aging for at least four months or after accelerated aging for 7 days at 70 degrees centigrade. (No label claim or expiration date is involved or allowed for this minimal data.)
20.0 Other Claims Requiring Data:
List any other claim that needs data to support it. ____________________________________
___________________________________________________________________________
___________________________________________________________________________
20.1 List in a table format the appropriate assay and timeframe for the evaluation that you used for each of the claims in 20.0.
21.0 510(k) Summary/Statement Requirement: (See Chapter 7, Summary and Statement Information.)
You MUST include on a SEPARATE sheet(s) your name, address and either a:
You may not begin commercial distribution of a device in the United States until you receive a letter from FDA stating that your surgeon’s glove was found to be substantially equivalent. Marketing the device prior to a finding of substantial equivalence would render the device adulterated under §501(f)(1)(B) of the FD&C Act and would be subject to enforcement action by the FDA.
Dynamic System
FDA Quality System Regulation (diagram)
Quality System & QS Audits (diagram)
Quality System Documents *
Design and Development Planning
Design Control System Outline
Design Input *Documenting Design Output
Acceptance Criteria
Design Output ApprovalDesign Verification and Validation
Labeling Verification
Design Transfer
Design Changes
Design History File
Sample Design Input Requirements Procedure *
Written Procedures
Sample Device Master Record Glove Specification
Record Retention *
COMPONENTS AND MANUFACTURING MATERIALS
Component Qualification
Specifications
Supplier Evaluation
Acceptance Procedures
Acceptance Criteria
Testing and Inspection
Acceptance and Rejection Records
Obsolete, Deteriorated, and Rejected Product
Product Storage
Receiving History Record
Receiving Latex Test Data
Component and Material Status Decals
Contamination Control
Environmental Control
Monitoring
Personnel Practices
EQUIPMENT AND MANUFACTURING MATERIALS *
Maintenance, Inspection, and Adjustment
Manufacturing Materials
Control Use
Receipt and Release
Area Separation and Inspection
Storage
Packaging
Procurement, Acceptance and Storage
Packaging Process
Sample Label for Dispenser Box
Shipping for Processing
PRODUCTION AND PROCESS CONTROL
Specifications
Processing ControlsPinholes
Processing Chemicals
Water-Soluble Proteins
Bioburden ControlReworking
Retesting
Sample Lab Analysis of Latex Compounding
Example Machine Dep Line Parameters Per Day
Ethylene Oxide (EtO )
Radiation Sterilization
Contract Sterilization
Complaint Handling System
Complaint Responsibility
Complaint Records
Investigation Records and Location
Medical Device Reporting *
Sample Complaint Processing Procedure
CORRECTIVE AND
PREVENTIVE ACTION
CHECKLIST FOR DESIGN
OF NATURAL RUBBER LATEX GLOVES AND PROCESSES
Manufacturers of medical gloves are required to meet the Good Manufacturing Practices (GMP) regulation for medical devices in 21 CFR 820. The GMP regulation requires that every manufacturer of finished medical devices shall prepare and implement a quality system that is appropriate to the specific device manufactured and meets the requirements of the regulation. Because of the requirement for a quality system, the GMP is called a Quality System regulation.
A network as shown in the diagram below is based on input, functions or tasks to be performed, output, and feedback. A network becomes a system only when it is correctly established, managed and operating, the paths are connected, and information is fed back and used to correct any problems in the system or the output. Thus, a system is self-correcting and is called a dynamic system because it functions as if alive.
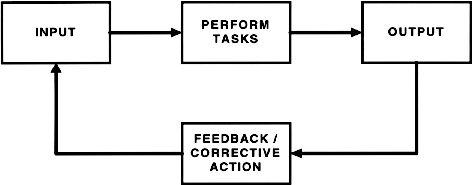
The medical device good manufacturing practices regulation requirements for a quality system covers objectives and policies for management and continues with the review of the quality system, quality related activities and corrective and preventive action. The required quality system contains several general elements or tools, particularly in §§820.05 to 820.25, to guide management and support the system. These management elements/tools are outlined next so that their extent can easily be seen.
__________________________
1The illustrative examples, procedures and forms included in this manual
are for educational purposes only. They show one method, but not the only
method, for performing a quality system function. Do not use these examples,
procedures and forms without first modifying them to meet your specific
requirements, operations and devices. Please see the disclaimer on page
iv.
Management Responsibility and Related QS Elements
| 820.20(a) | Quality Policy |
| 820.20(b) | Organization |
| 820.20(b)(1) | Responsibility and Authority |
| 820.20(b)(2) | Resources |
| 820.20(b)(3) | Management Representative |
| 820.20(b)(3)(i) | QS Established and Maintained |
| 820.20(b)(3)(ii) | Report QS Performance to Management With Executive Responsibility |
820.20(c) |
Management Review |
| 820.20(d) | Quality Planning |
| 820.20(e) | Quality System Procedures |
An overview of the required quality system is shown below in the diagramed FDA Quality System Regulation. (This is the same type of dynamic system as diagramed above. Note that a system diagram becomes more complex as the functions in each box are detailed.) The QS diagram below clearly shows the role played by all levels of management and by feedback and corrective action. For example, an audit may be directed by management or be triggered by data flowing through the corrective action path. Briefly, the QS requirements, as diagramed from top to bottom, cover:
In day-to-day activities, the QS functions are interconnected as needed to design, produce and distribute safe and effective gloves and to take any needed corrective or preventive action (CAPA). The next diagram, Quality System and QS Audits, expands the overall system diagram to show most of the broad functions covered by the QS regulation. This diagram shows many paths between the various functions. For example, 14 paths are shown into, and 12 paths out of, employee (personnel) controls to other areas. (Please note that it is not feasible for such diagrams to show all possible functions and interconnecting paths.)
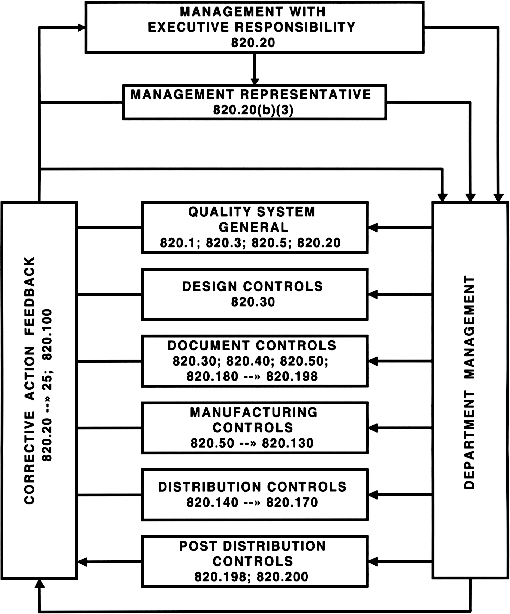
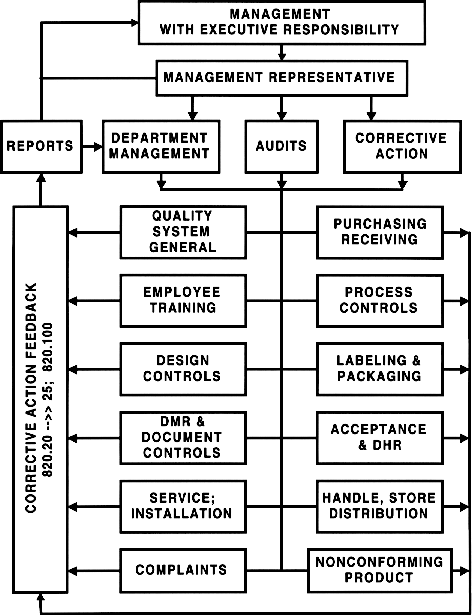
Each device manufacturer must prepare and use QA procedures adequate to assure that a formally established and documented quality system is implemented. "Established" means defined, documented, and implemented (per 820). Meeting the system requirements and this definition requires an obvious commitment to quality by management as shown by policy statements; assignment of responsibilities and authorities; and actions that define and support the quality system. Manufacturers must have the records required by the QS regulation such as:
Employee training is described in more detail in Chapter 5, Personnel, of the Medical Device Quality Systems Manual: A Small Entity Compliance Guide available at: http://www.fda.gov/cdrh/qsr/05prsnl.html Training requirements apply to all employees that perform any function covered by the Quality Systems regulation.
It is not unusual for FDA investigators to conduct factory inspections and see employees who are unaware of situations that can result in poor product quality. An example is glove strippers who are not paying attention to the condition of the formers. They have not been properly instructed to identify formers with charred latex, cracks, or chips; and give the former number to the line supervisor. These employees have not been properly advised that formers with these conditions can cause defects in gloves.
FDA employees have also seen compounders who have not been instructed on the importance of thorough ball milling to prevent particles with incorrect sizes from entering the dipping tanks. The ball milling master record may state to grind for 20-24 hours or 28-32 hours; but has the important nature of this operation been told to the compounders to assure that they follow the written instructions? Employees have been seen sweeping floors near coagulant and latex dipping tanks. We have seen employees handling gloves while wearing sharp-edged rings or other jewelry. We are always advised that it is the manufacturer’s policy not to allow jewelry or to require finger cots over ringed fingers; but are employees periodically reminded of the reason for the no ring requirement and why dipping tank solutions and formers must be kept clean?
The QS regulation requires in §820.25 that each manufacturer have sufficient personnel with the necessary education, background, training and experience to assure that all operations are correctly performed. Employees must be made aware of glove defects which may occur from improper performance of their specific jobs. Also, quality assurance or other verification personnel must be made aware of defects and errors likely to be found in defective components, gloves. Some defects are not visible such as micro-holes, high bioburden, excessive moisture, adverse chemical residues and high protein levels.
Personnel who perform verification and validation activities must be made aware of defects and errors that may be encountered as part of their job functions.
Proper job performance by employees in accordance with the QS regulation requires management that has a good knowledge of the QS regulation. Therefore, management including marketing managers who may receive complaints or review corrective action and preventive action (CAPA) should also have appropriate education, background, training and experience.
Quality assurance employees should meet the QS personnel requirements stated above for manufacturing employees and should be made aware of defects and errors likely to be found in defective components and gloves. Also, appropriate QA employees should be made aware of defects that can result from contaminants such as manufacturing materials, debris, charred starch, and moisture. Usually it is easier to teach all of the QS personnel requirements to all employees.
In order to meet the proactive requirements in §§820.25 and 820.100, management should diligently look for factors that indicate a need for additional training or retraining. This information is derived from management observations, analysis of device history records, analysis of complaint records, and quality assurance audits. Some of these factors are:
Design controls cover the practices and procedures that are used to help assure that the design of a glove is safe and effective, and meets the intended use, user/patient needs, applicable regulations, and applicable standards. The details of design control systems vary depending on the complexity of the product or process being designed. However, manufacturers of surgeon's gloves are expected to define, document and implement design control procedures as required by the QS regulation. FDA has proposed that patient examination gloves be reclassified as class II. If patient examination gloves are reclassified, then design controls would also apply to them. Design Controls are in §820.30 and are listed below:
(a) General,
(b) Design and development planning,
(c) Design
input,
(d) Design output,
(e) Design review,
(f) Design
verification,
(g) Design validation,
(h) Design transfer,
(i) Design
changes, and
(j) Design history file.
These design controls are shown below in the diagram, "Design Control System Outline." This diagram is an expansion of one element, design controls, from the system diagram shown earlier. (This overview design control diagram cannot show all paths nor show the number of times each path is used.)
Manufacturers may establish one design control procedure to cover the various design control sections; or, they may use one or more procedures for each topic. Multiple procedures may be easier to develop, update and implement. General design control procedures may be part of the quality system records or files noted in §820.186.
Personnel training in §820.25 applies to employees that perform any activity covered by the QS regulation including design. Most technical employees need various amounts of training in device regulations, safety, risk analysis, labeling, human factors, verification, validation, design review techniques, etc. Manufacturers are required to establish procedures for identifying training needs and making certain that all personnel are trained to adequately perform their assigned responsibilities. Design personnel must be made aware of glove defects which may occur from the improper performance of their specific jobs. In particular, personnel who perform verification and validation activities must be made aware of defects and errors that may be encountered as part of their job functions.
Design and Development Planning
Developing and producing a new glove are very complex tasks. Without thorough planning, program control, and design reviews, these tasks are virtually impossible to accomplish without errors or leaving important aspects undone. Planning and execution of the plans are complex because of the many areas and activities to be covered. Some key planning activities are:
Each manufacturer shall establish and maintain plans that describe or reference the design and development activities and define responsibility for implementation. The plans shall identify and describe the interfaces with different groups or activities that provide, or result in, input to the design and development process. The plans shall be reviewed, updated, and approved as design and development evolves.
The plans should be consistent with the remainder of the design controls. For example, the design controls require a design history file (DHF) per §820.30(j) that contains or references the records necessary to demonstrate that the design of the glove was developed in accordance with the approved design plan and regulatory requirements.
One of the first elements in each design plan should be how the manufacturer plans to meet each of the design control requirements for the specific glove the manufacturer plans to develop; that is, the design plans should support all of the required design control activities. Such plans may reference quality system procedures for design controls in order to reduce the amount of writing and to assure agreement. Each design control plan should be broad and complete rather than detailed and complete. Broad plans:
Regardless of the effort in developing plans, they usually need updating as the ongoing development activities dictate. Thus, the QS regulation requires in §820.30(a) that the plans shall be reviewed, updated, and approved as the design and development evolves. The details of updating are left to the manufacturer; however, design review meetings are a good time to discuss and review changes that may need to be made in the design development plan.
Interface. Design And Development Planning §820.30(b) states: the plans shall identify and describe the interfaces with different groups or activities that provide, or result in, input to the design and development process.
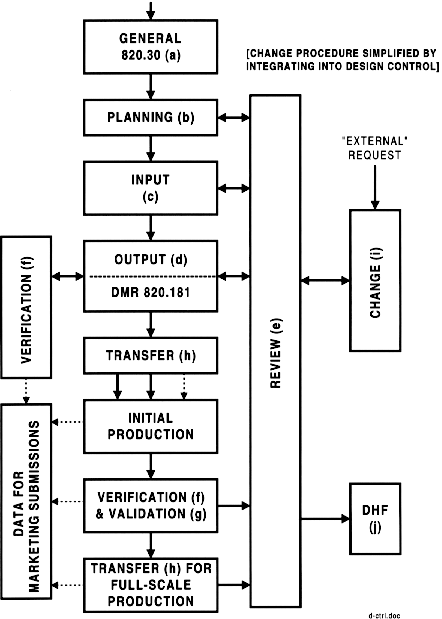
If a specific design requires special raw materials, clinical trials, support by another company facility or support by contractors such as performing a special verification test, etc., then such activities should be included or referenced in the plan and implemented in order to meet the interface and general quality system requirements. Of course, the interface and general requirements also apply to needed interaction with manufacturing, marketing, quality assurance, or other internal functions.
Because the development and manufacture of gloves is manufacturing process dependent, the interface between the device development and process development staff is extremely important and should be addressed in all of the general design control procedures.
Design input means the physical and performance requirements of a device that are used as a basis for device design per §820.3(f).
Design input, requires that each manufacturer shall establish and maintain procedures to make certain that the design requirements relating to a device are appropriate and address the intended use of the device, including the needs of the user. FDA considers shelf life or expiration dating to be a significant factor in meeting the needs of the user. Thus, shelf life should be considered under design input as part of the activities to meet user/patient needs. Also, FDA has published a proposed rule proposing that labeling contain an expiration date. FDA is further proposing that patient examination gloves be reclassified as class II after which they would be subject to design controls. Also, a design requirement in §820.130 requires that each manufacturer shall make certain that device packaging and shipping containers are designed and constructed to protect the device from alteration or damage during the customary conditions of processing, storage, handling, and distribution.
The design input requirements shall be documented and shall be reviewed and approved by a designated individual(s). The approval date and signature shall be documented.
Design input includes determining customer needs, expectations and requirements plus determining regulatory, standards, and other appropriate requirements. These requirements are documented by the manufacturer in a set of device requirements. The design input stage usually is a continuum because input requirements activities usually occur near the beginning of the design feasibility stage and continue to the early physical design activities. After the initial design input stage there are also activities to reduce the input requirements to engineering specifications. A set of design input requirements, when converted to engineering terminology, finalized and accepted as part of the device master record is called a device or product specification, or, in this case, a glove specification.
As the concept of the new or modified glove design is established, various user, patient and/or intended use questions should be answered. The questions and answers will vary for different types of gloves and accessories. Some typical broad and basic design input questions are:
Glove and process requirements and specifications should be reviewed and approved before physical design and process development begins to help control and direct all activities and increase the probability of achieving desired safety and performance characteristics. As the design evolves, the glove design, packaging, labeling, etc., shall be verified per §820.30(f) and reviewed per §820.30(e) against their specifications to verify that design input requirements have been met.
Glove requirements should identify all of the desired performance, physical, safety and compatibility characteristics of the proposed glove and, ultimately, the finished glove. Design input also includes requirements for labeling, packaging, and manufacturing. The final glove specifications should cover ALL of the glove characteristics. The glove specifications may incorporate other specifications by reference such as the manufacturer's generic list of specifications for a type of glove, or specific paragraphs or all of a standard, etc. It should be very clear exactly what is going to be met. A failure to properly address characteristics or factors such as biocompatibility (chemicals and proteins), barrier integrity, aging, packaging protection, shipping stability, reliability (expiration date), etc., can have disastrous consequences for barrier devices.
Input Checklists. It is possible to diligently develop glove requirements and still forget elements in the final specification. To reduce the probability of a requirement or characteristic being left out, a specification checklist(s) or general design list may be used during the design input stage. A checklist should be developed that is broad based but germane to the product line of the manufacturer. If used, a checklist should be part of a standard operating procedure such as a Design Input Procedure. A sample two-page general design and process control checklist is located at the end of this chapter.
The input requirements should cover applicable standards such as the glove standards by the:
American Society for Materials and Testing (ASTM).
100 Barr Harbor Drive
West Conshohocken, Pennsylvania 19428 USA
Phone: 610-832-9500 FAX: 610-832-9555
Information about most national and international standards may be obtained from the American National Standards Association (ANSI), 11 West 42nd Street, New York, New York, 10036, phone 212-642-4900.
The design input procedures must address incomplete, ambiguous, or conflicting requirements. Thus, every reasonable effort should be made to collect all of the requirements. Then the designers can review them and generate detailed design specifications that are clear, correct and complete. The design input requirements shall be documented, reviewed and approved by a designated individual(s). The approval date and signature shall be documented.
To the extent feasible, glove specifications should be derived from the input requirements and be documented before beginning the design of the actual glove. The glove and other related specifications should be kept current as the design of the glove, packaging, labeling and manufacturing processes evolve during the development program. As the physical design evolves, the specifications usually become more specific and detailed. The specifications will undergo changes and reviews as the design evolves. However, one goal of market research and design reviews is to establish complete glove requirements and specifications that will minimize subsequent changes.
Old versions of the input requirements and subsequent input specifications are put in the design history file (DHF) or indexed in the computer as part of the DHF to help show that the design plan was followed. The final specifications are part of the device master record.
Design review [§820.30(e)] is one of the key design control elements in a quality system. As the design and production processes evolve, design reviews reduce errors, help avoid problems, find existing problems, propose solutions, increase producibility and reduce production transfer problems. The relentless inquiry during design reviews will expose needed design input requirements and/or design corrections that otherwise may have been overlooked. Design reviews help assure that the final design of the glove, labeling, packaging and processes meets the current design requirements and specifications. Please see Chapter 3 of the Medical Device Quality Systems Manual: A Small Entity Compliance Guide.
Design output per §820.3(g) means the results of a design effort at each design stage and at the end of the total design effort. The finished design output is the basis for the device master record. The total finished design output consists of the glove, its packaging and labeling, and the device master record. The device master record (DMR) as defined in §820.3(j) means a compilation of records containing the procedures and specifications for a finished device.
The output at each stage is the documents and physical design elements that are either complete or are used to move the design into the next stage. For example, the first design output will usually be the design requirements documents from which the designers will derive the preliminary design specifications. Then the physical design begins including the selection of known components and raw materials and begin documenting their purchasing and acceptance requirements. Section 820.50(b), Purchasing Data, requires that each manufacturer shall establish and maintain data that clearly describe or reference the specified requirements (i.e., a specification), including quality requirements, for purchased or otherwise received product and services. Other components and raw materials will be selected as the design evolves. The design output for some special or new components, or components in unusual applications, will include verification protocols and data; and also include subsequent purchasing and acceptance requirements.
Many of the design output documents are directly part of the DMR. The remaining DMR documents are created by quality assurance, production or process engineering, technical writing, etc., using design output data and information. For example, the finished glove final-test methods and data forms may be derived from the design verification protocol(s). When these design and documentation activities are completed, the DMR is complete. When the DMR is complete and initial production units, including packaging, meet all specifications, the complete finished design output exists. The requirements in §820.30(d) contain three parts (numbers added) as follows:
Documenting Design Output (1) Documenting design output in terms that allow an adequate evaluation of conformance to design input requirements (or the requirements converted to specifications) is a significant design activity. A matrix or index of input specifications may be compared with the outputs to assist in assuring conformance. Another common technique for achieving conformance is listed below.
Each document has a different drawing number but the paragraph numbers are the same for each parameter. The first document generated may be copied and used as the format for the next one. Therefore, it is almost impossible to leave out a design parameter. When verification is performed and documented, conformance or lack of conformance from input specification to output documents and to output specification data is obvious.
Acceptance Criteria (2). The verification (discussed below) documents and data contain more information than is typically needed for production evaluation and acceptance of components such as latex, in-process items and finished gloves. Therefore, it is easy to copy and modify verification documents to meet the quality system requirement that:
Deriving production test procedures from the verification protocols also yields the DMR test methods and data forms needed to meet the QA procedures and acceptance criteria in §820.181(c). Some test methods such as for protein and powder exist as ASTM or other national or international standards. These may be used where appropriate.
Design Output Approval (3) The third output requirement is that design output shall be documented, reviewed, and approved before release. The approval, including the date and signature of the individual(s) approving the output, shall be documented. This means that:
Design Verification and Validation
Each manufacturer shall establish and maintain procedures for verifying the device design. Design verification shall confirm that the design output meets the design input requirements and associated specifications. The results of the design verification, including identification of the design, method(s), the date, and the individual(s) performing the verification, shall be documented in the DHF.
Verification means confirmation by examination and provision of objective evidence that specified requirements have been fulfilled. The design of gloves, and packaging and any subsequent changes should be verified by testing. Verification test data covers parameters such as shelflife (expiration date), tensile strength, elongation, pinhole AQL, protein residues, chemical residues, biocompatibility, correct labeling, etc. Because a major part of the glove specifications are usually derived from final or draft consensus standards, this means that the manufacturer determines by standard test methods that the gloves meet appropriate standards, guidance and any other claim established by the manufacturer for the gloves.
Design verification is done before final design validation [some validation may be done throughout the development program.] Design verification is always done against input specifications and validation is done against input requirements.
Verification and validation should be done by skilled personnel using test equipment calibrated and controlled according to quality system requirements. Verification and validation should be done per written protocol(s) that includes defined conditions for the testing. Protocol(s) may not be perfect, particularly a new design. Therefore, verification and validation personnel (with authority to made changes) should carefully annotate any changes to a protocol. Likewise, comments about any deviations or other events that occurred during the testing, use or simulated use should be recorded. The slightest problem should not be ignored. During design reviews, the comments and any deviations may be as important as verification test data
Design validation means establishing by objective evidence that device specifications conform with user needs and intended use(s). Validation follows successful verification testing and analysis. Each manufacturer shall establish and maintain procedures for validating the device design. Design validation shall be performed under defined operating conditions on initial production units, lots, or batches, or their equivalents. Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions… The results of the design validation, including identification of the design, method(s), the date, and the individual(s) performing the validation, shall be documented in the DHF. Validation may include use under real or simulated conditions to assure that the users are satisfied with the donning ability, feel, size, shape, texture, holding ability, tactile sensitivity, lack of fatigue, lack of irritation, color, odor, etc., of the gloves.
Appropriate laboratory and animal verification (performance, reliability, biocompatibility, etc.) testing followed by analysis of the results should be carefully performed before clinical testing (a validation test) or commercial distribution of the gloves. The manufacturer should be assured that the design is safe and effective to the extent that can be determined by scientific tests and analysis before clinical testing by humans or routine use by humans. Clinical testing is not needed for gloves substantially equivalent to gloves legally marketed in the United States.
Gloves manufactured for use in clinical studies under an IDE are exempt ONLY from the production section of the GMP/QS regulation. They are not exempt from design controls listed in §820.30. In addition, the IDE regulation has labeling requirements in §812.5 and quality assurance requirements in §812.20(b)(3) that shall be met. Further, manufacturers should remember that human subjects are protected via informed consent requirements and product liability laws.
Labeling should be checked to see if it is directed to the user and not to the glove designers, which is a common fault found in labeling. Text should be short and to the point yet transfer the maximum information to the user. Data, identifications, or other key information should be current, complete, unambiguous, and accurate. Note that much of the labeling text for gloves is specified by regulations and guidance. During verification, labeling is checked against the labeling requirements of the manufacturer, standards and FDA. Any instructions should be followed exactly by the verification test operators and such action should result in correct use of the glove. A checklist may be used to aid in the review of labeling.
The design controls require that each manufacturer shall establish and maintain procedures to ensure that the device design is correctly translated into production specifications [§820.30(h)].
A significant part of the transfer requirement is met when the design output is properly created. That is, many of the design output documents are part of the DMR. The remaining DMR documents are based on design output information. A procedure may be needed to cover the generation of the remaining DMR documents. Employee training, as needed, should be covered by the design transfer procedure. Design transfer should assure that the design being transferred:
The design output is transferred for initial production and validation. If problems occur, changes are made per change control procedures including design controls for Class II gloves. The transfer is complete when the finished gloves are validated, and all requirements are met.
Changes to a design element are controlled per §820.30(i) Design Changes which states that: each manufacturer shall establish and maintain procedures for the identification, documentation, validation or where appropriate verification, review, and approval of design changes before their implementation. However, the original design activities and subsequent change control activities for the design are both done under the full set of quality system design controls. For example, a major problem may result in additional design inputs, design planning, etc. An easy method for design change control is to use a change request procedure in conjunction with the regular design control procedures. This method reduces the number of procedures, amount of learning, and errors because the change control work is done using the regular design control procedures.
As the design activity progresses toward the final stage and as more items are approved, it is expected that the degree of change control will increase. Those elements of the design that have been verified and accepted should be under change control. Elements that have been released need to be under change control in order to develop production processes. A design that has been submitted to FDA for marketing clearance should be under change control. A design that is released for production must be under design control §820.30 and general document change control §820.40.
After the physical design evolves into an approved and accepted glove, subsequent changes to the glove specification(s) are to be proposed, evaluated, reviewed, approved, and documented per §820.30 [not just 820.30(I)]. The revised specification(s) becomes the current design goal in accordance with the manufacturer's procedures for design control, design change control, and document control. An overall design change control procedure should cover:
Design history file (DHF) means a compilation of records which describes the design history of a finished device [§820.3(e)]. The DHF covers the design plan, procedures and activities used to develop the device, accessories, major components, labeling, packaging and production processes. The design controls in §820.30(j) require that each manufacturer shall establish and maintain a DHF for each type of device. Each type of device means a family of gloves that are manufactured according to one DMR. Documents are never created just to go into the DHF.
The QS regulation requires that the DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of this part. This requirement cannot be met unless the manufacturer develops and maintains plans that meet the design control requirements. The plans and updates should be part of the DHF. In addition, the QS regulation specifically requires that the results:
The DHF is not required to contain all design documents or to contain the DMR, however, it will contain historical versions of key DMR documents that show, to some extent, how the design of the glove, labeling, packaging, and processes evolved. Typical documents that may be in, or referenced in, a DHF are listed below:
Sample Design Input Requirements Procedure
A sample Design Input Requirements procedure is presented below which covers basic activities for obtaining requirements that are needed to develop glove specifications. This procedure uses the multiple specification approach; however, a single combined specification would use a similar procedure. This procedure should be modified to meet specific needs before being adopted.
| Sample procedure. Do not use without modifying to meet your specific needs |
| C O M P A N Y L O G O |
Sheet 1 of 2 | |
| Title: Design Input Requirements Procedure | SOP Number | |
| Prepared by: | Date Prepared | |
| Approved by: | Date | Rev |
| ECN notes | ||
POLICY - Design specifications covering all design requirements shall be established for all proposed glove designs before any significant physical design activities are started.
SCOPE - This policy applies to all gloves and accessories developed by our company or developed by a contractor for us. For purchase of completed designs, refer to SOP ####. The glove specification(s) must exist or be generated regardless of the source of the initial design.
CONFIDENTIALITY - Development plans and activities are confidential. Market research reports and documents such as specifications with parameter data shall be marked confidential.
Design control procedures, standard SOPs, and required design review and design verification/validation records may be shown to, and copied by, FDA investigators as required by the QS regulation. Design parameters are not covered by the QS regulation. Therefore, confidential specification characteristics and parameters in the copies of documents shall be blacked out unless the document is being collected during an inspection related to a marketing submission.
RESPONSIBILITY
Marketing and Engineering have the primary responsibility for determining safety and performance requirements and developing input specifications; however, all departments are expected to support the development of input requirements and subsequent specifications.
MARKETING - Marketing shall plan and conduct all customer contacts to obtain information on customer desires, needs, expected pricing, opinions about existing gloves, etc.
To the maximum extent feasible, market research shall be conducted in a manner to reduce leaking of manufacturer confidential information and plans.
Design review meetings shall normally precede and follow all significant outside market research activities. Initial market research activities shall be previewed with top management.
Market research results are to be documented and marked confidential.
PRODUCTION - Production engineering has primary responsibility for assuring producibility and establishing manufacturing requirements. Some of these requirements may be general during the early design stages. (Process development is also done under design controls.)
R&D ENGINEERING - R&D Engineering is expected to supply design input information on most requirements. Such inputs may parallel data obtained by market research.
R&D has primary responsibility for specifying what technology to use.
R&D shall analyze input data on requirements and reduce it to preliminary specifications.
R&D has primary responsibility for addressing incomplete, ambiguous, or conflicting requirements and shall see that such issues are appropriately discussed at design reviews.
RA & QA - RA and QA managers or their designees shall attend all design input or specification review meetings to provide input on, and to assure that, regulatory, company, quality, safety, performance, etc., procedures are followed and that requirements are met.
SPECIFICATIONS
STRUCTURE - Multiple specifications shall be used. A separate specification shall be developed for accessories, labeling, packaging, etc. An overall glove specification shall be developed and shall include an index that points to supporting specifications. The specifications, among other factors, shall address:
CHECKLISTS - Checklists of requirements germane to our product line may be used to develop and support specifications. If used, such checklists become part of this procedure and part of the design documentation.
DESIGN REVIEW - Each glove specification shall undergo design review before it is approved for physical design activities or is used as a background document to support further market research. Such reviews shall be documented.
APPROVAL - The Marketing manager and R&D Engineering manager shall approve all input specifications after these have been subjected to design review.
DOCUMENTATION - The approved specifications shall be given document numbers and become part of the device master record for the new glove.
CHANGE CONTROL - The Engineering manager shall decide when design activities have progressed to the stage that the various specifications shall be subject to our Design Change Control Procedure. Approved items that have been released for use shall be under change control. Design change control can start no later than the submission of a 510(k).
*****End of Procedure*****
A device master record (DMR), required by §820.181, is a term used in the QS regulation for all of the design output and related documentation required to manufacture a glove. A DMR contains documents for typical manufacturing activities such as procurement, processing, labeling, test and inspection, and packaging. The device master record contains the design, formulation, specifications, complete manufacturing procedures, quality assurance requirements or acceptance criteria, packaging and labeling of a finished glove. Almost all sections of the QS regulation have requirements related to the DMR. The device master record is described in Chapter 8 of the Medical Device Quality Systems Manual: A Small Entity Compliance Guide.
A device master record for medical gloves manufactured by complex processes such as latex dipping usually contain many documents. For convenience, many manufacturers generate an index or table of contents which lists all of the documents in a device master record. An index is a valuable list which increases a manufacturer’s state-of-control, reduces costs by reducing the effort and time required to locate a document, and helps manufacturers meet the QS accessibility requirement for records. Also, a matrix, table or diagrammed DMR index may be used to help show that design outputs fulfill design input requirements per §820.30(d). The DMR should contain a glove (device) specification. Generally a medical glove specification will include the:
Many sections of the QS regulation require written procedures for guidance in performing various design, QA and manufacturing tasks. Written procedures are used to:
Medical gloves tend to require a relatively large number of written procedures because of the lack of visual clues and complex nature of the manufacturing processes. Written procedures and history records are needed, for example, for mixing of latex, coagulant, dipping, leaching, cleaning, and other solutions. A written procedure is needed for the collection, storage, accelerated testing and real-time evaluation of finished gloves to establish shelf life or expiration dating.
*** SAMPLE RECORD *** This is an example of an examination glove specification including some typical parameters. You may modify this form and use it to meet your needs. Do not use this example as is.
| C O M P A N Y L O G O |
Sheet 1 of 1 | |
| Title: Glove Specification | SOP Number | |
| Prepared by: Crystal Thompson | Date Prepared 8-1-95 | |
| Approved by: Althea Barcome | Date 8-8-97 | Rev C |
| ECN notes: ECN 429 Protein limit added 8-1-96; no label claim -- no new 510k required; ECN 436 Rev B powder level changed to 120 mg per glove | ||
| ECN 438 Rev C protein reduced to 1200 µg per glove by process improvement | ||
COMPANY PRODUCT NAME: Crystal Touch | |||
| Trade Name: | Patient examination gloves, non-sterile | ||
| Intended Use: | Medical activities except surgery | ||
| U.S. FDA Status: | Class I, Classification Number 80LYY;
510(k) marketing clearance required. Must be manufactured under quality system program. | ||
Material: |
Natural rubber latex |
||
| Donning Lubricant: | U.S.P. Absorbable corn starch | ||
| Catalog Numbers: | Crystal Touch 100S | | Crystal Touch 100M | | Crystal Touch 100L |
| Sizes: | Small | | Medium | | Large |
| Overall Length: | 240 mm minimum | | 240 mm minimum | | 240 mm minimum |
| Width: | 80 +/- 10 mm | | 95 +/- 10 mm | | 111 +/- 10 mm |
| Palm Thickness: | 0.10 mm minimum | ||
| Finger Thickness: | 0.10 mm minimum | ||
| BEFORE AGING | | AFTER AGING | ||
| Tensile Strength: | 22 Mpa minimum | | 17 Mpa minimum | |
| Ultimate Elongation: | 750% minimum | | 550% minimum | |
Orientation: |
Ambidextrous |
||
| Cuffs: | Yes | ||
| Color: | Natural | ||
| Residual Powders | 120 mg max. per glove | ||
| Protein Max. | 1200 µg per glove max. | ||
Packaging: |
100 units in dispenser box by weight |
||
| Disp. Carton Labeling: | Per Packaging specification P-192 Rev. C | ||
| Ship. Case Labeling: | Per Packaging specification P-193 Rev. B | ||
| Product Coding: | |||
The QS regulation in section 820.180 requires that all records pertaining to a device be retained at least two years from the date of release for commercial distribution or for a period of time equivalent to the design and expected life of the device. For most medical gloves the longer of two years or the labeled expiration/shelflife date, is an adequate period for retaining records.
COMPONENTS AND MANUFACTURING MATERIALS
The QS regulation requires that both components and manufacturing materials be addressed by the manufacturer’s quality assurance program. More information is described in Chapter 10 of the Medical Device Quality Systems Manual: A Small Entity Compliance Guide.
Components, raw materials, manufacturing materials, etc., are called products in the QS regulation. Components and materials are products used during manufacturing which are intended to be part of the finished gloves. Examples of such products are: the latex concentrate; the various chemicals which will be mixed with the latex or polymer during compounding; and the donning lubricant. Labeling and packaging are specified and procured the same as components. Manufacturing materials are substances used to help in the manufacturing process but are not intended to become part of the finished gloves. Examples include: most of the ingredients in the coagulant dipping solution, any detergents that are used to clean the formers, and starch if the starch is added and then removed to make "powder-free" gloves.
Components, labeling, packaging, and manufacturing materials must:
The quality program should assure that the incoming product is:
Qualification consists of verifying through documented testing, evaluation, and review that a product will reliably perform its function in the intended application per §820.30(f) (that is, a component or material meets the specifications derived from the glove input requirements).
Components and other materials should be selected using the requirements of the finished glove as a guide. Standard products that are used in their normal application will need only minor testing unless some characteristic such as chemical allergenicity was not adequately covered in past studies. New products such as donning lubricants, protein modified latex or new accelerators usually need testing. A design history file of any qualification (verification) testing of components, raw materials and manufacturing materials must be maintained for surgical gloves. This record should include the product identity, testing methods that were used, who performed the testing, date, and the actual test data and results.
Component and material specifications are required to be part of the device master record. The specifications should adequately describe the characteristics, dimensions, design, materials, viscosity, performance, and any other feature or parameter necessary to assure receipt of the product desired. For standard products that have a known performance history, a catalog designation may be adequate to describe a component or material and assure purchase of the desired product. For unusual, new, or very important components or materials, the specification data is derived primarily from the qualification data plus minor details from catalog data. Specifications should reflect design requirements, quality and reliability needs. For some components, such as donning powder, the specifications should include a limit on micro-organism contamination.
To the extent feasible, the selection of suppliers of services and product and the evaluation of them by audits, performance analysis, etc., should be part of a quality program per §820.50. If the manufacturer does not have the capability to test certain product for conformance to specifications, then supplier test data or outside lab results are acceptable per §820.80. Any outside test results should be for the specific lot received and should be accompanied by relevant raw data so that a judgment of authenticity may be made by the finished glove manufacturer.
Section 820.80 requires a written procedure for accepting components, raw materials, manufacturing materials or other product. Before being accepted, all incoming products should be either physically separated (quarantined) or clearly identified as not yet accepted per §820.86. The decision to separate or tag not-yet-accepted products should be made based on the characteristics of the components, materials, manufacturing materials and gloves; the potential for mixups; plant conditions; manufacturing practices; etc.
To the extent technically feasible, manufacturers should have specific acceptance criteria for components, materials, manufacturing materials, etc., to meet §§820.30, 820.181, and 820.50. Acceptance criteria are the attributes of a product that determine its acceptability, such as appearance, color, dimensions, MST, percent solids, preservative levels, protein levels, viscosity, purity, pH, or performance characteristics. Typically, acceptance criteria are part of the inspection and test procedure or may be in a separate document.
All incoming components, materials, or other product should receive at least a visual inspection for contamination and/or damage and be identified as the product specified on the purchase order. Product must be tested if deviations from specifications could result in medical gloves being unfit for the intended use. Any testing and inspection must be performed according to written procedures in order to meet the documented QA or QS program requirements and written acceptance test requirements.
Manufacturers who decide not to sample or test selected product should be able to justify that decision based on such factors as knowledge of the supplier’s previous performance in providing quality product, the product performance history, and application of the component, material or other product. If product is tested by the supplier, acceptance of incoming product can be based on certification and review of test data submitted by the supplier for the specific components, materials, etc., supplied. Certification should accompany each lot of product. When certification is used, the manufacturer should periodically verify the validity of the certification through an audit of the supplier, testing of the received product or other means.
If a contract laboratory is used to test components, materials, manufacturing materials, etc., the laboratory becomes an extension of the glove manufacturer’s quality system. The glove manufacturer is responsible for assuring that the contractor’s test and inspection procedures and quality system are acceptable. Typically, this assurance is obtained by documented audits.
Acceptance and Rejection Records
The QS regulation specifies that a record of product acceptance and rejection be maintained per §820.80. These records are a part of the device history record (DHR) and should be maintained in a format that will help in the review of the history record. The records are not required to be maintained in a single file with other records, and are typically filed in the receiving or quality control area. Typically, acceptance and rejection records should contain:
Obsolete, Deteriorated, and Rejected Product
Occasionally a lot of gloves or product will not meet specifications. Typically, defects in chemicals, polymers, and finished gloves are not visible. Therefore, it is very important that containers of rejected gloves and product and obsolete and deteriorated product be identified per §820.86; and, these should be placed in a separate quarantine area or specially identified area to prevent mixups. To assure that unacceptable products are not used, §820.90 requires that records of their disposition be maintained. These records should state whether the gloves, components, materials, or other products were returned or scrapped.
When components, raw materials, manufacturing materials, etc., are not immediately processed, such product should be held in a quarantine area or identified upon receipt as not yet accepted per §§820.86 and 820.150. Components, raw materials, manufacturing materials, etc., must be identified or stored so that their status is obvious; that is, the material has been accepted, rejected, or is awaiting a disposition decision. A quarantine area can be either a physically secure area or simply a limited access area identified as a quarantine area.
Components should be stored such that they are protected from moisture, dirt, and insects. Components or materials such as latex and starch must be stored so as to retard the growth of micro-organisms §820.70(e). Where components and materials degrade, the components must be stored in a manner to facilitate stock rotation such as first-in, first-out use per §820.150. Ceramic formers and other items not affected by the environment may be stored in sheds or outside.
| If you use any sample procedures or forms in this manual, you should modify them to meet your specific needs. | |
| *** SAMPLE RECORD*** |
sheet __ of _ |
| Description of Product: | Test/Inspection* Procedure #: | |||||||
| Purchase Order # |
Lot # |
Date Rec’d |
Supplier | Lot Size |
Quantity Accepted |
Quantity Rejected |
Inspect Date |
Signature |
* The acceptance activities and test equipment are listed in the test and inspection procedures for each product.
*** SAMPLE RECORD *** |
|
| Do not use without modifying to meet your specific glove and operations. | |
| SUPPLIER: | Date Received | |
| SOURCE: | Date Tested | |
| LATEX GRADE: | Lot | |
|
PARAMETER |
PROCEDURE NO.* |
RESULTS |
| % TOTAL SOLID CONTENT | ||
| MST (sec) | ||
| % DRY RUBBER CONTENT | ||
| % NON-RUBBER CONTENT | ||
| % ALKALINITY ( NH3% ) | ||
| VFA NUMBER | ||
| KOH NUMBER | ||
| VISCOSITY | ||
| pH VALUE | ||
| OTHER: | ||
| Remarks: *Acceptance activities and test equipment to be used are stated in the respective test procedures listed above. | ||
| Tested by (signature): | Time | |
*** SAMPLE RECORD Status labels or decals (next page) to help meet §§820.80 and 820.86 in the QS regulation. Note that these sample records are different from those previously published for the 1978 GMP regulation.***
COMPONENT AND MATERIAL STATUS DECALS (Samples)
(RIR = receiving inspection report) |
|
QUARANTINED |
RIR |
|
| |
|
APPROVED |
RIR | ||
| Product or Material | |||
| Part or Spec # |
Quantity | Signature | Date |
| Remarks
| |||
|
REJECTED By Quality Control | |||
| Product or Material
| |||
| Part or Spec # |
Quantity | Signature | Date |
| Remarks
| |||
Buildings in which components, raw materials, manufacturing materials and finished gloves are handled, processed, and stored should have sufficient space and be designed to allow proper cleaning, maintenance, and other operations. There should be adequate space for receiving, manufacturing, packaging, labeling, and storage to minimize contaminates, assure orderly handling procedures and prevent mix-ups. Buildings should be designed and arranged so that operations can be performed in an orderly manner and thus reduce confusion that can lead to unsatisfactory job performance and mix-ups. Different operations or processes should be separated either by walls or partitions or by providing enough space between operations to preclude mix-ups and assure that no activity will emit spray or dust, or otherwise have an adverse effect on adjacent activities. Some raw materials and gloves with different sizes, formulations, characteristics are not readily identifiable by sight. Therefore, orderly operations, product status, identification, etc., are very important to prevent product mix-ups. Buildings and environment are also described in Chapter 6 of the Medical Device Quality Systems Manual: A Small Entity Compliance Guide.
The need for contamination and environmental control during the collection, compounding and processing of latex is well known. Studies by FDA of micro-photographs have shown that particulates are often associated with pinholes and weak spots in gloves. Thus, for each area in the building where components and gloves are processed, any element such as dust, paint chips, rust, starch residues, protein residues, microorganisms, humidity, temperature, static electricity, etc., which a manufacturer has determined might cause contamination should be controlled. Buildings should be appropriately constructed to prevent, reduce, and control these conditions and support the manufacturer’s environmental control program. For example, the control of dust may require that driveways and parking lots be paved. Floor sweeping and floor polishing scatter dust which can contaminate wet formers, coagulant dipping tanks, latex/polymer dipping tanks, finished gloves, etc. Therefore, floors must be cleaned by washing or other dust reducing methods. Floor polishers should not be allowed in glove factories. Likewise, equipment should be cleaned as needed by methods that will reduce or prevent contamination of tanks, formers and gloves with dust and other debris.
The lack of environmental control will result in the contamination of materials, formers, dipping tanks, wet product, etc. Some environmental factors to be considered are lighting, ventilation, temperature, humidity, static electricity, insect and pest control, and particulates such as dust, rust, paint chips, grease drips and starch. If rodenticides, insecticides, etc., are used, written procedures to limit their use and for their removal from work surfaces should be established to prevent any adverse affect on the manufacturing process or the device per §820.70(c) and (e). For example, finished gloves, solution tanks, etc. should be covered during application of pesticides to the room.
Each manufacturer should make prudent decisions as to what environmental controls are necessary to assure the quality of gloves made by their particular process. When determining the control needed, the manufacturer should identify exactly what needs to be controlled, such as:
Packaging and starch should be stored in a clean, dry, insect-free area. Latex should be preserved to prevent bacterial growth. Components such as starch that support bacterial growth should be stored in a controlled environment such as sealed containers or bags. Unfiltered air should not be used to dry washed formers or coagulant-coated formers as the resulting contamination may cause pinholes. Unfiltered factory air should not be used to cool or dry finished surgeon’s gloves. Open windows and doors should be screened to control insects.
An appropriate system for regular monitoring should be established and maintained for the environmental factors to be controlled. This will assure that equipment is performing properly and filters, floors, equipment, etc. are clean, and other aspects of the environment are within specifications. Periodic inspections of environmental controls and documentation and review of the inspections are required by §820.70(c). The inspection record should be kept simple.
Adequate bathroom, dressing, storage and waste facilities should be provided, as appropriate, for personnel to maintain cleanliness per §820.70(d). Such facilities should be maintained on a scheduled basis. Where necessary, such as in a controlled room for inspecting and packaging surgeon’s gloves, special clothing and an area to don and store these garments may be needed. Clean area clothing should not be worn into uncontrolled rooms or outside per §820.70(e) because the clothing will become contaminated.
Eating and smoking create particulates that may cause pinholes in medical gloves. Smokers exhale particles up to 15 minutes after they finish smoking. These activities should be confined to designated areas. Also, containers or equipment should be provided for timely and safe disposal of trash, by-products, effluents and other refuse per §820.70(e).
EQUIPMENT AND MANUFACTURING MATERIALS
The QS regulation requires that all equipment used to manufacture a device be designed, constructed, placed and installed to facilitate maintenance, adjustment, cleaning, and use. The degree of maintenance of equipment and frequency of calibration of measuring equipment will depend on the type of equipment, frequency of use, and importance in manufacturing processes.
Manufacturing materials such as mold release compounds, cleaning agents, lubricating oils, and other substances used to facilitate manufacturing are procured and received the same as components. If any of these materials has an adverse effect on the finished glove, then the adverse material must be removed to an amount that does not adversely the quality of the glove using an approved written procedure per §820.70(h). Equipment, manufacturing materials and calibration are described in Chapter 7 of the Medical Device Quality Systems Manual: A Small Entity Compliance Guide.
Maintenance, Inspection, and Adjustment
Glove formers should be automatically cleaned each cycle by a validated process. Formers should be inspected as gloves are stripped or as appropriate. Defective formers should be lifted, or removed and repaired, or replaced. Because dust, grease, charred starch, paint chips, etc., can cause pinholes and other defects in medical gloves, manufacturers must maintain formers and maintain, clean, protect and adjust processing equipment. Therefore, manufacturers should:
The proper or optimum operation of manufacturing equipment and the operation of a dipping processes usually require the use of manufacturing materials. The QS regulation in §820.3(p) defines "manufacturing material" as any material or substance used in or used to facilitate the manufacturing process, a concomitant constituent, or a byproduct constituent produced during the manufacturing process, which is present in or on the finished device as a residue or impurity not by design or intent of the manufacturer. "Concomitant constituent" means naturally occurring material and for natural rubber latex includes latex proteins. Manufacturing materials also include latex or polymer processing chemicals that are not intended or desired to be in the finished gloves. Manufacturing materials are included in the definition of product in §820.3(p) and are specified, procured, inspected and/or tested, etc., the same as other products such as components per appropriate requirements in §§820.30, 820.50, 820.181 and 820.80. The Quality System regulation in §820.70(h), Manufacturing material states:
Where a manufacturing material could reasonably be expected to have an adverse effect on product quality, the manufacturer shall establish and maintain procedures for the use and removal of such manufacturing material to ensure that it is removed or limited to an amount that does not adversely affect the device’s quality. The removal or reduction of such manufacturing material shall be documented.
The use of manufacturing materials that may adversely affect the finished glove should be carefully analyzed. Each process should be designed to use a minimum amount of adverse chemicals so as to reduce costs, reduce removal efforts, and increase the safety of the glove.
The QS regulation in §820.70(h) requires a written procedure for the use and removal of manufacturing materials that can have an adverse effect on devices. For medical gloves, processing of raw latex and leaching and washing processes are commonly used to remove or denature natural water-soluble proteins and remove adverse materials such as processing chemical residues. Manufacturers should develop, validate, document and control latex processing and leaching and post-cure washing or treating processes to assure that the finished medical gloves meet their (the manufacturer’s) specification for residual or trace powder level, chemical residues, and water-soluble proteins or specific allergens, as appropriate. When residues from sterilization agents such as ethylene oxide need to be removed, instructions for aeration are necessary.
Where starch is added to medical gloves to expedite handling and is then removed during further processing, for example, during the production of some "powder-free" gloves, the starch becomes a manufacturing material. A written processing and test/inspection procedure should be used to assure that powder residues on the finished gloves meet finished device specifications.
Glove manufacturers must include labeling and packaging in their QS program per §§820.120 and 820.130. Labeling includes dispenser box labels, case labels, package labels, and directions for use such as caution statements about the removal of starch from gloves after donning. The QA program should assure that labeling meets the device master record requirements with respect to readability and content and assure that labeling operations are controlled so that the correct labeling is always issued and used. Labeling and packaging are also described in Chapters 11 and 13 of Medical Device Quality Systems Manual: A Small Entity Compliance Guide.
Specifications are required in the device master record for the content and design of labeling. Specifications are the engineering drawing and/or artwork for each item of labeling, and the appropriate inspection and control procedures. The drawings or purchase specifications should specify, as appropriate, the label dimensions, ink, finish, and content so that the purchased label and/or printed packaging will remain legible during the customary conditions of processing, storage, handling, distribution, and use; and contain the correct and intended claims. Labeling claims must match glove characteristics and contain required labeling in order for the gloves not to be misbranded (see chapter 6 on labeling.) All procedures, drawings, and artwork must have the name of the author, an approval signature, and a date. The approval signature and date may be on the back of artwork or on a labeling approval form. Labels and printed packaging are specified and purchased as components.
Before transfer for initial use, labeling should be reviewed and approved by marketing, quality assurance, and other appropriate managers. Manufacturers should have a design control procedure which covers the drafting, review, and approval of labeling. Approval forms are generally used in conjunction with such a procedure. This procedure helps prevent misbranding.
Labeling is part of the device master record; therefore, all changes to labeling must be made under a change control system. Changes must be formally reviewed and authorized before use according to §820.30 for surgeon’s gloves and per §820.40 for patient examination gloves. [If gloves become Class II as proposed, then patient examination gloves must also be subject to §820.30].
Upon receipt, packaging and labeling materials must be examined and, if deemed necessary by the manufacturer, tested to assure conformance with specifications per §§820.80 and 820.120. A designated individual(s) shall examine the labeling for accuracy, where applicable, the correct expiration date, control number, storage instructions, and any additional processing instructions. After being accepted, these labels and packaging materials may be placed into inventory or into production. This release, including the date and signature of the individual performing the examination, must be recorded in the device history record. These activities should be repeated when labeling is removed from storage and released for use in production.
Area Separation and Inspection
All labeling and packaging operations being performed at the same time should be separated as necessary to assure there are no mixups between similar labels or various sizes of gloves. Gloves with different protein levels should have a high degree of separation. Separation may be either a physical separation or by performing the labeling and packaging at different times for different types or sizes of gloves. Before beginning any packaging and labeling operation in which mixup could occur, the production area and equipment should be examined to make certain that any gloves and labeling materials from previous operations have been removed. Unused labeling that contains a pre-coded manufacturing date, expiration date, or lot number, should be destroyed and not returned to the label storage area.
Preprinted packaging and labeling materials should be stored in a suitable area and manner to prevent mixups. Labeling or prelabeled glove dispenser boxes should be identified and segregated as necessary to prevent mixing of similar labeling. Access to labeling should be limited to authorized personnel.
An effective primary package for a medical glove should be designed and developed along with the product by considering glove characteristics, contamination control, sterilization process, sealing, labeling, secondary packaging, shipping, environment, shelf life (expiration date), end use, and FDA regulations per the design controls in §§820.30 and 820.140. The primary package and the shipping container should adequately protect gloves under all reasonable conditions from original packaging to final use. Complete storage and stability data should be compiled for packaging for sterile gloves or obtained from the supplier of the packaging.
The process capability of packaging and sealing equipment for sterile gloves should be determined and documented. Then a sealing cycle should be selected. Because every package is not tested or inspected, the cycle must be validated, and then documented in a setup and operations procedure to be used for routine packaging and sealing of the glove with the selected packaging materials. Finally, manufacturers should perform quality assurance tests and inspection on samples of the finished packages to further assure that company specifications are met.
Procurement, Acceptance and Storage
The device master record should contain appropriate specifications so that the desired packaging, labels and components may be purchased, properly stored, and properly used. Primary packaging for gloves to be sterilized should be kept clean before sterilization. A manufacturer should have adequate procedures for approval or rejection of all incoming adhesives, packages, cartons, etc. Suppliers may test and inspect these components and provide the manufacturer with the results for each batch (i.e., certificate of conformance). The manufacturer could accept this specific data as sufficient certification or order his own testing and inspection.
The packaging operation is a manufacturing process. Therefore, the QS sections for processing controls (§§820.70 and 820.75) and finished device inspection (§820.80) apply to packaging operations. Controls should be adequate to assure that labeling is correct for the package contents and that only gloves approved for release are packaged and released. Released gloves should meet the manufacturer’s specification for dryness (moisture content). It is very important that starch-coated gloves be dry because starch supports microbiological growth.
Section 820.181(d) requires that the device master record include packaging and labeling specifications, methods and processes. Written instructions should be provided to assure that the necessary controls are understood and consistently used. Manufacturers should have a written procedure for test and/or inspection of finished packages. The packaging of sterile gloves should be tested and/or inspected before and after sterilization for integrity; and such testing and inspection is usually done on a sampling basis. The results of test and/or inspection should be recorded in the device history record along with lot numbers, if any, because §820.184(d) requires records which demonstrate that the device was manufactured in accordance with the DMR requirements.
*** SAMPLE *** For training purposes only. Do not copy. Always check labeling requirements in 21 CFR 801. |
| MANUFACTURER’S LOGO | Drawing Number: 301-443-6597 | Rev C | ||||
| USE: Exam Gloves | Title: Dispenser Box | Sheet 1 of 1 | ||||
|
Approved Larry Andrews |
Date 2/5/89 |
| Size: | See drawing below. | Material: | Fiberboard weight__ gms/m2 |
| Closure: | Suitable for Acme No. 123 Hot Melt or equivalent. | ||
| Style: | Rectangular box with round end dispenser top slot 1" wide x 5" long. | ||
| Application: | For convenient dispensing of examination gloves. | ||
| Folding: | Scored for folding. | Shipping: | Ship flat. |
| Printing: | Lettering - Carolina Blue Background - Arctic White |
Type Font - Bold | |
| Size: | Minimum ¼" for "LATEX EXAMINATION GLOVES" Minimum 3/16" for other printing | ||

Either a quarantine area or a label control system must be used to prevent distribution of gloves marked "sterile", but not yet sterilized. The required level of control is high.
If the labeled gloves are to be shipped to a contract sterilizer, the shipping, handling, and processing must be controlled as required by the QS regulation and §801.150(e) of the labeling regulation. Section 801.150(e) is reprinted below.
(e) As it is a common industry practice to manufacture and/or assemble, package, and fully label a device as sterile at one establishment and then ship such device in interstate commerce to another establishment or to a contract sterilizer for sterilization, the Food and Drug Administration will initiate no regulatory action against the device as misbranded or adulterated when the non-sterile device is labeled sterile, provided all the following conditions are met:
(1) There is in effect a written agreement which:
(i) Contains the names and post office addresses of the firms involved and is signed by the person authorizing such shipment and the operator or person in charge of the establishment receiving the devices for sterilization.
(ii) Provides instructions for maintaining proper records or otherwise accounting for the number of units in each shipment to insure that the number of units shipped is the same as the number received and sterilized.
(iii) Acknowledges that the device is non-sterile and is being shipped for further processing, and
(iv) States in detail the sterilization process, the gaseous mixture or other media, the equipment, and the testing method or quality controls to be used by the contract sterilizer to assure that the device will be brought into full compliance with the Federal Food, Drug, and Cosmetic Act.
(2) Each pallet, carton, or other designated unit is conspicuously marked to show its non-sterile nature when it is introduced into and is moving in interstate commerce, and while it is being held prior to sterilization. Following sterilization, and until such time as it is established that the device is sterile and can be released from quarantine, each pallet, carton, or other designated unit is conspicuously marked to show that it has not been released from quarantine, e.g., "sterilized—awaiting test results" or an equivalent designation.
Compliance with §801.150(e) may require two written agreements when importing pre-labeled "sterile" but not-yet-sterilized surgeon’s gloves. The two agreements are:
Where the contract sterilizer and manufacturer or importer are located in the same state, a written agreement such as described by §801.150(e) will also satisfy the QS status requirements in §820.86 and some of the handling requirements in §820.140 for shipments between the person authorizing shipment and the contract sterilizer.
Gloves that have been sterilized and shipped to the manufacturer’s warehouse before final release must be properly labeled. Pallets, or other designated units, must be marked to indicate the status of the gloves, such as "sterilized awaiting test results". The manufacturer should be able to show that it has control of the gloves until final release and, if necessary, could have them destroyed or returned for reprocessing.
A 510(k) for surgeon’s gloves should be submitted by the person having direct or contractual control over the sterilization. If the manufacturer does the sterilization or contracts for the sterilization, the manufacturer submits the 510(k).
For imported prelabeled "sterile" but not-yet-sterilized surgeon’s gloves, the importer that contracts for the sterilization should submit the 510(k) to the FDA. However, the manufacturer of the gloves should be identified in the submission.
Change control is of the utmost importance and is described in detail in Chapter 9 of the Medical Device Quality Systems Manual: A Small Entity Compliance Guide. Inadequate change control:
Change control applies to all QS elements. For example, change control applies to:
Design change control requirements for gloves are covered at the beginning of this chapter under Design Controls. The production documentation and document change control requirements are in §820.70(b) and §820.40 Document Controls and require that each manufacturer shall establish and maintain procedures to control all documents that are required by this part. The procedures shall provide for the following:
(a) Document approval and distribution. Each manufacturer shall designate an individual(s) to review for adequacy and approve prior to issuance all documents established to meet the requirements of this part. The approval, including the date and signature of the individual(s) approving the document, shall be documented. Documents established to meet the requirements of this part shall be available at all locations for which they are designated, used, or otherwise necessary, and all obsolete documents shall be promptly removed from all points of use or otherwise prevented from unintended use.
(b) Document changes. Changes to documents shall be reviewed and approved by an individual(s) in the same function or organization that performed the original review and approval, unless specifically designated otherwise. Approved changes shall be communicated to the appropriate personnel in a timely manner. Each manufacturer shall maintain records of changes to documents. Change records shall include a description of the change, identification of the affected documents, the signature of the approving individual(s), the approval date, and when the change becomes effective.
A change control procedure and associated forms is one of a family of SOP’s used to produce, number, select drawing size, change, and control documentation. The production change procedure must describe the manufacturer’s approved procedures to be followed from the time the device master record is first released for production of a glove, or a change is requested for a glove design or manufacturing processes, through review of the change in relation to other appropriate documents, activities, and use. [Design change control before transfer to production is also required for surgeon’s gloves. See §820.30 and the beginning of this chapter.] The change procedure should be flexible because all changes do not need the same degree of evaluation and approval. However, all changes MUST be made according to the manufacturer’s procedure. Making uncontrolled changes is a very serious violation of several sections of the QS regulation.
Each changed glove; component such as colorants, flavors, odorants, antimicrobials, stabilizers, antioxidants, lubricants, etc.; and labeling, packaging, or process must be thoroughly evaluated and reviewed by appropriate personnel to verify that manufacturing specifications are met. Specifications include allergenicity parameters such as residue levels for adverse manufacturing materials, primary skin irritation and dermal sensitization. Thus, the need for allergenic studies should be considered when raw materials, manufacturing materials and processes are changed. Then the test results and all information related to the change should be reviewed by the change-control board (review group). [This procedure covers the same activities as needed for introducing a new product or process into production.] The change control procedure should state the details of the evaluation and review process. The procedure should define the responsibilities of the various departments and members of the change-control board.
*** SAMPLE PROCEDURE ***
| C O M P A N Y L O G O Title Change Control Procedure Prepared by ______________________________________ Approved by Date Rev ______________________________ |
Sheet 1 of 3 |
| ECN Notes ________________________________________________________________ | |
PURPOSE AND SCOPE: To establish a procedure and form for requesting, evaluating and approving changes. This policy and procedure covers all of our gloves intended for distribution.
POLICY: It is our policy that any change to the glove design/formulation, manufacturing, labeling and packaging must be evaluated, reviewed and approved before the change is made. This procedure is used in conjunction with our design control and process validation procedures.
FORMS: Engineering Request (ECR) Form and Change Order (ECO) Form. The form is a request form until the proposed changed is approved.
REVIEW BOARD: Proposed changes and accepted changes shall be reviewed by a group composed of at least the Engineering, QA, and Production Managers. (Manufacturer Type A approval) If a glove, labeling or packaging change is proposed, Sales and marketing shall participate in the ECR review. (Manufacturer Type B Approval)
If the proposed change is estimated to have a significant cost impact, the Plant and Finance Managers shall be informed. (Manufacturer Type C Approval)
The Engineering Manager shall be the chairperson of the Review Board.
RESPONSIBILITY: Engineering has the major responsibility for managing the change control and review process.
Identification: The person proposing the change must complete the top of the ECR form and check the ECR block. State the item to be changed, date, drawings, and other appropriate information. Give the ECR to Engineering for an initial review.
ECR Review: The Review Board shall review ECR’s and decide if a requested change will be accepted, and developed into a change order (ECO).
Effective Date: The Review Board shall set the processing date and/or shipping date for the first lot of gloves processed under a specific change order.
Responsibility: The Engineering Manager, with guidance from the Review Board, shall decide which department or designee is responsible for each task to be performed in order to develop the change, evaluate it, and implement it.
Evaluation: Glove performance, barrier properties, allergenicity and biocompatibility must be considered after each change. After the changed glove, label, process, etc., is developed and tested, the results must be thoroughly verified and evaluated by appropriate departments. Process validation must be considered and performed if needed. Then the test results and all information related to the change shall be reviewed by the change-control Review Board. The meeting notes plus the change-control forms, drawings, or other appropriate documents shall record the details of the change. The revision level letter on each changed drawing or procedure shall be increased by one letter. For example, from Rev. A to Rev. B. (During initial production, the revision codes will be numbers; the example changes from Rev. 1 to Rev 2.)
Documentation Distribution: Engineering shall distribute the revised device master records to the persons responsible for the operations affected by the change and shall remove old documents and file or discard them, as appropriate. Supervisors are responsible for overseeing the use of new device master record documents, especially if a change is being phased in and the old and revised documentation are being used in their department at the same time.
Disposition of In-Process Items: The Change Review Board is responsible for any special instructions for the disposition of old products. The Manager affected by the change shall be responsible for the disposition of in-process items such as labeling and packaging that cannot be used after the change.
Premarket Notification: The Change Review Board shall determine if a 510(k) needs to be submitted for, and cleared before, the change may be implemented. "YES" or "NO" decisions shall be documented. (Some manufacturers may have a Regulatory Affairs person to assist with these decisions.)
Remedial Actions: The Change Review Board and complaint handling personnel, if appropriate, shall be responsible for advising the Plant Manager if a recall is proposed.
Quality Assurance Review: After the change is implemented, QA personnel must make certain that finished gloves meet the specifications in the revised Device Master Record by:
| *** SAMPLE RECORD *** |
Sheet 3 of 3 |
| MANUFACTURER LOGO | Engineering Change Order (ECO) |
ECO # | ||||
| Signatures | Date | Approvals | Date | Reason for Change | ||
| Originator | Type A | [ ] Improve process [ ] Biocompatibility [ ] Design improvement | ||||
| Project Engr. | Type B | [ ] Correct error [ ] Cost reduction [ ] Customer request | ||||
| TYPE
A[ ] B[ ] C[ ] |
Type C | [ ] Labeling or packaging [ ] Regulatory | ||||
| DESCRIPTION OF CHANGE | Action Code |
Drawings Affected | REVISED | |||
|
From |
To | |||||
| CHANGE ACTION REQUIRED | CODES | CHANGE ACTION REQUIRED | CODES | |||
| Purchasing and Production |
Scrap in-process products | 1 | Bioburden control | 8 | ||
| Rework finished gloves | 2 | Labeling | 9 | |||
| Repackage finished gloves | 3 | Packaging | 10 | |||
| Re-sterilize finished gloves | 4 | Other Action |
For reference only | 11 | ||
| Notify supplier | 5 | Employee training | 12 | |||
| Allergen control | 6 | 10(k) required for Change |
13 | |||
| future | 7 | 510(k) not required | 14 | |||
PRODUCTION AND PROCESS CONTROL
During production, all of the factors covered by the QS regulations should be considered in order to control the manufacturing process and produce safe and effective medical gloves. The objective of production and process control is to assure that:
Specifications describe the intent of the design, and processes are planned so that gloves produced by them meet these specifications. For a given design, note that these specifications are the glove-specific documents in the device master record. Of course, general documents in the device master record are also used to manufacture gloves.
The QS regulation requires manufacturers to establish procedures to ensure that the DMR is prepared and approved in accordance with §820.40. Because surgeon’s gloves are listed in 820.30(a), the DMR is prepared per §§820.30 and 820.40. The DMR is adequate if the design configuration and performance requirements can be consistently met when the gloves are manufactured and packaged according to the DMR specifications for raw materials, compounding, processing, quality tests and inspections, packaging and labeling, etc.
Manufacturers must establish process controls to insure that the gloves are not adversely affected by the process and that the process will achieve its intended purpose. Process controls include process validation, standards, drawings, written procedures and instructions, monitoring, in-process glove evaluation, operator certification, finished glove inspection and test, etc. The number of written procedures to assure process control depends on the nature and complexity of the process and the training of the operator(s).
Manufacturers should assure that all processes are conducted properly by controls such as training, supervision, audits, inspection, testing, documentation, automated processes, etc. All changes to processes must be properly reviewed, validated, documented, and communicated to appropriate employees in a timely manner.
The physical examination and testing of all in-process and all finished gloves for all parameters is impractical with present technology. For example, only a small statistical sample of gloves is usually tested for pinholes; and allergen tests are usually done on a design basis with infrequent follow-up tests. Therefore, gloves need indirect control by process validation, sample testing, and subsequent monitoring of the processing methods, equipment, and personnel. A sample two-page general design and process control checklist is located at the end of this chapter. Such processes must be developed, validated, documented, and controlled such that the finished gloves consistently meet the manufacturer’s glove specifications. Guidance on process validation is on the web at: http://www.fda.gov/cdrh/comp/ghtfproc.html
Pinholes. Studies by FDA of micro-photographs of defective latex gloves have shown that dust, dirt, rust, paint chips, charred starch, insect parts, and other debris are often associated with pinholes. Therefore, appropriate environmental and processing controls are needed to reduce debris on wet formers and in compounding and dipping tanks. Unfiltered air should not be used to dry wet formers where the resulting dusty/dirty former will go into a coagulant or latex/polymer dipping tank. Floors and equipment must be cleaned by methods that minimize the amount of debris released into the environment. As feasible, dipping tanks should be protected from environmental debris. Equipment components, construction materials, rollers, paint, etc., should be selected to reduce initial and long-term production of debris. Likewise, rollers, bumpers, parts, etc., that could shed debris into dipping tanks should be replaced before they become heavily oxidized or degraded. (See the Appendix for a list of labs that perform water-leak testing.)
Excessive grease may fall on formers or into tanks and cause defective gloves. Also, only the very thin film of grease at the contact surface lubricates. Therefore, the amount of grease should be limited on chains and equipment moving over formers and processing tanks.
Starch and charred starch in the environment are debris and need to be controlled. Obviously, the amount of starch released into the environment needs to be minimized. Cleaning methods such as washing should be used that will remove starch without further scattering it into the environment. (Also see the section on Environmental Control in this chapter.)
Manufacturers should control other causes of pinholes such as vibration, air bubbles in the dipping tanks, dirty formers, defective formers, incorrect formulation, exessive curing temperatures, too little dewebber, wet coagulant, etc. These controls are necessary because manufacturers should use controls and processes that, to the maximum extent feasible, produce finished gloves that meet the manufacturer’s specifications and regulatory requirements. Inspection and testing of finished gloves to separate good gloves from defective gloves is not acceptable to FDA as the primary means of process control.
Processing Chemicals. Coagulating and other processing chemicals that are not intended to be on the finished gloves are manufacturing materials. These must be reduced on the finished device below the adverse level per §820.70(h). For gloves, the level is set such that the gloves will meet the manufacturer’s quality claims and regulatory requirements. Leaching, washing, surface treatment or other processes for removing adverse manufacturing materials should be developed, validated, documented and continuously controlled in order to meet the established device master record specifications. Suitable test methods should be developed and validated for routine testing of samples of finished gloves to assure that manufacturing material residue levels are met. If used, any secondary test method should be validated versus appropriate chemical or other standard primary test method. Also see http://www.fda.gov/cdrh/ode/944.html.
Water-Soluble Proteins. Water-soluble proteins and manufacturing materials on latex gloves have been implicated in the literature and in adverse incidents as the cause of allergic reactions. Processing controls include the reduction of adverse manufacturing materials and water-soluble proteins. Of course, the control would be the reduction of specific allergens if all of such allergens are known. If standard or regulatory recommendations or limits exist, they should be met. (See Labeling in Chapter 6.)
Current investigations and conventional manufacturing techniques indicate that one way of minimizing such reactions is to remove as much of the water-soluble proteins and adverse manufacturing chemicals as is feasible from latex gloves. This removal is primarily done by:
Washing after curing is important because proteins become more water-soluble and/or move to the surface of latex gloves during heat curing. Thus, washing should be done before the final donning powder or lubricant, if any, is applied. Otherwise, the starch slurry tank is saturated with water-soluble proteins. Chemical residues, protein on the surface of the gloves, and protein in the slurry tank become attached to, or coat, the starch and other particulates. Later, some of the particulates with residues and protein could become airborne during handling and use of the gloves.
The temperature of the leaching and washing water should be established by each manufacturer as the temperature needed varies based on the parameters of the overall compounding, dipping and curing methods. Preliminary studies by the Malaysian Rubber Research Institute and others indicate that the purity (flow rate) and agitation of the leach water and total leaching time are more important than water temperature.
Surface treatment of the cured latex glove with chlorine or similar agents denatures surface constituents such as water-soluble proteins. These treatment processes also wash and rinse away proteins and manufacturing residues. Chlorine is an adverse manufacturing material and must be removed per §820.70(h) from the gloves after chlorination by washing, neutralization, etc
Synthetic polymer gloves, polymer-coated latex gloves, or any gloves with a labeled or controlled protein level should not be dipped in any tank (particularly starch slurry tanks) or tumbled in dryers where regular protein coated latex gloves have been produced unless the tanks are cleaned before the production of the low- or non-protein gloves. Otherwise, such low- or non-protein containing gloves may become contaminated with protein.
The processes used to control water-soluble proteins and manufacturing materials must be developed per §820.30, validated per §820.75, documented per §820.181, thereafter controlled per §§820.70 and 820.75 and operated by trained personnel. Validation guidance is available on the web at www.fda.gov/cdrh/comp/ghtfproc.html.
A suitable method should be used for sample testing for water-soluble proteins and adverse chemical residues or specific allergens during routine production. Such methods should be validated versus standard laboratory methods. Data from validation of the testing methods for proteins and manufacturing materials or specific allergens should assure that the test methods are adequate. Use of these test methods in production should show that leaching, cleaning or treating processes being used adequately reduce water-soluble proteins, adverse manufacturing materials or specific allergens to, or below, the level set in the manufacturer’s specifications. (ASTM or other standards organization may establish protein, chemical, total extractables, and/or specific allergen levels in the future. Meanwhile, manufacturers should set their own levels which should be consistent with current practice and medical needs. Please see the FDA proposed recommendations for protein and powder in Chapter 6, Labeling.)
Bioburden Control. Medical gloves, particularly those powdered with starch, can support the growth of micro-organisms. Therefore, processing controls, as appropriate, should include:
An example which stresses the need to exercise controls over conditions related to microbial growth is demonstrated by a recall of examination gloves. It was the manufacturer’s practice to reduce the temperature of the glove drying oven when serious mechanical problems occurred. After experiencing a problem and restarting the operation, a lot of insufficiently dried gloves containing cornstarch was packaged in a moist state. After distribution, a hospital called FDA and complained that they had noted a visible black film on the surface of the lot of gloves, and that their analysis revealed cultures of Aspergillus and Fusarium. Fortunately the manufacturer had lot numbers on the product which were traceable to the date the moisture problem occurred, and thus were able to restrict the recall to specific lots of examination gloves.
Finished gloves must be evaluated according to written procedures to show that the lot of meet all of the manufacturer’s specifications (acceptance criteria) per §§820.80 and 820.181. The finished device evaluation must include inspection and testing of samples of completely finished gloves. Because of different flow/bleeding/leaking characteristics from pinholes, leak test procedures for synthetic polymer gloves may have to be different than for natural rubber latex. The gloves selected for testing, as appropriate, are powdered, powder-free, cured, post-washed, chlorinated, lubricated, packaged, sterilized, etc., such that they are the same as the gloves delivered to the user. Glove evaluation, as appropriate, covers parameters such as:
Evaluation of finished gloves is usually done on a sampling basis. The sampling level and sampling frequency for each parameter are not necessarily the same. Device evaluation also usually includes appropriate in-process inspection and testing. The evaluation data for a lot of gloves, etc., must be recorded in the device history per §820.80(e) and §820.184, and reviewed per §820.80(d) before the lot is released for distribution. Historical data may be used to tighten or loosen sampling plans. There is some concern that the number of pinholes increase as gloves age. Thus, glove aging during shipment and storage before use should be considered before any sampling plans are modified [§§820.250, 820.100, and 820.40].
Powder Measurement. The American Society for Testing and Materials (ASTM) D-6124 Standard Test Method for Residual Powder on Medical Gloves was published September 1997. FDA has accepted this standard as the method for measuring total residues (trace powder) on "powder-free" gloves. (ASTM is developing a method for measuring donning powder on powdered gloves and FDA is considering accepting this method when it is published. Currently, it is expected to be a second part of D-6124.)
If medical gloves fail to meet specifications for parameters that can be tested and/or inspected, the gloves may be 100 per cent tested and/or inspected to separate those that meet specifications. The QS requirements for reworking nonconforming product in §820.90 and finished device evaluation in §820.80 apply to these activities.
Specifications for reworking should document the specific tests and processes to be performed. These specifications should be based on studies that measure the effects of reprocessing operations. For process type industries, manufacturers should evaluate reprocessing to assure that gloves will not be adversely affected. The results of the evaluation should be documented. For example, following re-sterilization, gloves should be inspected on a sampling basis for characteristics which may have been altered. Some examples of effects that may need consideration are:
Gloves and components to be reprocessed must be identified to distinguish them from acceptable gloves and components per §820.86. Identification of these may be done, preferably by marking their containers, or by identifying the area in which they are held.
Manufacturers should implement appropriate QA checks (acceptance criteria) to assure reworked gloves meet specifications. When gloves are reworked, the gloves must be subjected to reinspection, and/or testing, as necessary to assure that the reprocessing was adequate and did not have an adverse effect on the performance of the gloves per §820.90(b)(2). In most cases the procedure(s) used to inspect and test the original gloves are adequate for reworking.
*** SAMPLE OF AN IN-PROCESS RECORD *** |
| DATE | SHIFT | |
| Batch Number |
RESULTS | |
| Compounding Tank No. | ||
| Date & Time Compounded | ||
| Date and Time of Test | ||
|
PROCEDURE* |
BEFORE | |
| 1. pH | ||
| 2. TSC | ||
| 3. CCl4 | ||
| 4. NH3 | ||
| 5. VISCOSITY | ||
| 6. OTHER | ||
|
PROCEDURE* |
AFTER MATURATION | |
| 1. pH | ||
| 2. TSC | ||
| 3. CCl4 | ||
| 4. NH3 | ||
| 5. VISCOSITY | ||
| 6. OTHER | ||
| Remarks:
*The test & acceptance activities performed & equipment used are described in the procedures | ||
| Tested by:
Signature: | ||
| *** SAMPLE OF AN IN PROCESS RECORD ***Do not use without
modifying to meet your specific needs.
MACHINE DIP LINE PARAMETERS PER DAY LINE: ___________________ SHIFT: _____________________ DATE:_______________ OPERATOR: _______________________________ SUPERVISOR:
__________________ |
|
ITEM OR PROCESS |
TIME & PARAMETER |
ACTION TAKEN | ||
| 1. Acid tank 55-600 C. | ||||
|
Level |
||||
| 2. Rinse 45-500 C. | ||||
|
Level |
||||
| 3. Water 55-600 C. | ||||
|
Level |
||||
| 4. Wash oven 80-850 C. | ||||
| 5. Coagulant 50-550 C. | ||||
|
Level |
||||
| 6. Coagulant oven 80-850 C | ||||
| 7. Latex dip 27-290 C. | ||||
|
Level |
||||
| 8. Tack oven 85-900 C. | ||||
| 9. Beading | ||||
| 10. Leaching 55-650 C. | ||||
|
(1) Level |
||||
| 11. Leaching 55-650 C. | ||||
|
(2) Level |
||||
| 12. Cure oven #1 temp. | ||||
| 13. Cure oven #2 temp. | ||||
| 14. Cure oven #3 temp. | ||||
| 15. Protein Rinse operating | ||||
| Chain speed | ||||
| Line start time | ||||
| Line stop time | ||||
| ~ items produced / hour | ||||
| Length of glove (23-24 cm) | ||||
| Weight of glove (gm) | ||||
| Reject weight (kg) | ||||
| Remarks | ||||
For gloves imported into and sterilized in the U.S., the 510(k) must be submitted by the importer, U.S. subsidiary of the foreign manufacturer, or other U.S. party having control over the handling, shipping and sterilization. The sterilization information required in a 510(k) for gloves that are labeled "sterile" should include the following:
Guidance on sterility is in ODE Bluebook Memo K90-1 510(k) "Sterility Review Guidance 2/12/90" which may be obtained from DSMICA by phoning Facts-On-Demand 301-827-0111 or 800-899-0381 and requesting document number 361 or obtained from our web site at: http://www.fda.gov/cdrh/k90-1.html
The physical parameter and biocompatibility data in a 510(k) submission should be data obtained from test and inspection of packaged and sterilized gloves or it will not be accepted by FDA. The original document (not a copy) of biocompatibility study results should identify the test laboratory and should be kept on file by the organization submitting the 510(k).
If the inner package of surgeon’s gloves is labeled "sterile" before sterilization, the shipping containers of gloves must be handled, labeled, shipped and sterilized as required by U.S. 21 CFR 801.150(e). These factors must also be controlled in order to meet Quality System requirements in 21 CFR Part 820.
Packaging, package seals, labeling (i.e., ink) and bioburden for gloves must be compatible with the intended sterilization process. Please see Bioburden Control in the preceding section on Production and Process Control.
The U.S. FDA seeks a sterility assurance level (SAL) of 10-6 for surgeon's gloves. That is, the sterilization process should be designed so that the probability of a glove being non-sterile is 1 in 1,000,000 even if the gloves originally contained highly resistant microorganisms.
Medical gloves must be sterilized with a validated sterilization process. Gloves sterilized during the validation runs may be distributed if all sterilization and performance specifications are met. A sample from each sterilization load of the packaged and sterilized gloves should be inspected for physical parameters and package integrity after sterilization. Gloves may be re-sterilized if the manufacturer has process validation data to demonstrate that re-sterilization will not degrade the product or packaging below the finished device specifications.
The lethality of EtO sterilization is usually monitored with certified biological indicators (BI’s) and by measuring process parameters. These BI’s usually consist of 106 (i.e., 1,000,000) Bacillus subtilis var. niger spores on a prepackaged strip or self contained spore strip and growth media. Before cycle validation, the load configuration for cartons of gloves to be placed in the sterilizer should be established and documented. Likewise, the location of BI’s should be specified. Usually BI’s are located in a geometric pattern that covers the entire chamber load with at least one BI located in the coldest location. The coldest location is determined by a heat distribution study after the sterilizer is calibrated and determined to be working correctly.
During process development, the packaged gloves in the shipping cartons with BI’s are placed in the chamber. The chamber is evacuated to remove air and steam is injected to heat and humidify. The EtO gas is then injected to expose the product load per the proposed process parameters for one-half of the proposed cycle. Then the BI’s and gloves are tested for sterility per the consensus requirements of the United States Pharmacopoeia (U.S.P., a private company) to determine if all organisms are killed or the time for total kill may be extrapolated from the fractional kill data. Usually three half-cycle runs are done during process development to make sure the time for total kill of the 106 BI spores and glove bioburden is correctly determined. The half-cycle is doubled for production on the basis that an additional 106 spores would be killed to yield a SAL of 10-6 (commonly called a SAL of 6). The three half-cycle lots may be re-run for a full cycle and distributed if all parameters including EtO residues are met.
Usually the first three production runs are considered to be validation runs and require sterility testing of the sterilized gloves, and extra operator attention, extra temperature monitoring, and BI monitoring to make certain that the production cycle yields consistent results. (See guidelines by Association for the Advancement of Medical Instrumentation (AAMI), 3330 Washington Blvd., Suite 400, Arlington, Virginia, 22201-4598, United States.) For routine production using the validated full cycle, exposure and testing of BI’s and process parameter measurements are sufficient for sterility assurance -- sterility testing of samples of the sterilized gloves is not required.
EtO sterilization leaves residues of EtO, Ethylene Chlorohydrin, and Ethylene glycol. Natural rubber has a relatively high absorption rate for EtO when compared to common plastics. The recommended residue limits are those for "devices contacting skin" as stated in the June 23, 1978, pg. 27482, U.S. Federal Register. Residue dissipation curves are generated during the development and validation runs. Aeration cycles are established such that the finished gloves will always have residues below the limit set by the manufacturer. Residue levels do not need to be measured for each production run.
Primary packaging for gloves to be sterilized with EtO must allow the rapid passage of air and EtO through the package to prevent blowouts and allow degassing while preventing the passage of microorganisms.
The primary packaging, adhesive, gloves, and sterilization dose must be designed or selected and verified per §820.30 so that the packaged product will not immediately or later be degraded below specifications by the radiation. [For sterile examination gloves the manufacturer performs these per their company requirements in order to meet their label claims in §820.181, Device master record.] Degradation studies should be conducted. Exposure to high temperature may be used for accelerated life testing but should be followed by real-time testing. The test plan and results must be documented per §§820.30 and 820.70.
If AAMI methods are used, the radiation dose to achieve sterility should be based on the bioburden of the packaged gloves. The gloves used for bioburden testing and establishing the dose should represent routine manufacturing conditions. To assure that the bioburden for routine production is as low as the levels used to establish the radiation sterilization dose, packaging materials and gloves should be kept clean throughout storage, handling, processing and post-processing handling, and storage until the packaged gloves are sterilized.
If a manufacturer decides to use a dose of 2.5 mega-rads (25KGy), AAMI method 3 may be used provided the bioburden of the packaged gloves does not exceed 100 colony forming units. When samples of packaged gloves are exposed to a verification dose of 4.4KGy, statistical verification is accepted if there is zero or one positive sterility sample observed. A glove sterilized by the over-kill method should meet an SAL of 10-6.
The packing density and load configuration should be established and documented. Then the load is dose mapped by the sterilizer. Previous dose mapping results may be used to reduce but not eliminate dose mapping for the specific product now being considered. Packing density and load configuration affect the radiation penetration of the product. If packaging density, glove thickness, or load configuration are significantly changed, the manufacturer or contract sterilizer should decide if revalidation of the product sterilization process is needed.
Gamma sterilization is usually monitored by dosimeters. Beta (electron-beam) sterilization is usually monitored by recording the beam current and other accelerator equipment parameters. The electron-beam dose is verified by dosimetry.
Production and contract sterilization of gloves must be performed such that the glove manufacturer and the contract sterilizer meet the applicable parts of the QS regulation and the labeling requirements in §801.150(e). (Please see the section on Shipping for Processing for details.)
Complaint processing is described in Chapter 15 of Medical Device Quality Systems Manual: A Small Entity Compliance Guide. Information that tends to be specific for gloves manufactured by dipping processes is presented in this section.
Complaints from all sources should be processed per the manufacturer’s complaint handling procedure. The manufacturer should assure that personnel in marketing, sales, engineering, manufacturing, etc., report complaints. These employees must be made aware of this QS requirement and this should be noted in their training records. Complaints may be received from:
Because gloves are a low cost device, user reporting for tears and leaks is estimated to be low. Thus, a small increase in the rate of complaints may be significant.
Employees that maintain complaint files and conduct complaint investigations should have a thorough knowledge of latex gloves in order to make an informed, reasonable decision as to the severity of a complaint and to decide if an investigation is necessary. If it is decided that an investigation is not necessary, a record must be made of the reason for, and the individual responsible for, this decision. For example, the complaint may be about another manufacturer’s product or the same as another recent complaint that has already been investigated and resolved.
Each manufacturer should establish a method for maintaining records of complaints and investigations that is easy to use, meets their needs and meets the requirements of the QS regulation. A form, usually two-sided, is commonly used to help process complaints. A computerized system may be used. One side or page is typically used to record incoming complaint information and the other side or page is typically used to record the investigation of the complaint. An example procedure and forms are shown below. These forms list typical data that may be received and information that may need to be sought in order to adequately document complaints and investigations for gloves.
Investigation Records and Location
An investigation may be triggered by individual defects or failure of a lot depending on the nature of the failure, the manufacturers acceptable quality level (AQL), claims about incorrect labeling, regulatory requirements or customer criteria.
In some cases the failed gloves may be available for an investigation of the mode of failure. Failure analysis should be conducted by appropriately trained and experienced personnel and may require the services of a test laboratories. Investigators should use written procedures to assure that handling and analysis of returned defective gloves will not destroy the evidence that may show the cause of failure. For example, washing contaminated gloves will destroy evidence about chemical and protein residues. The failure investigation and analysis should determine the actual problem or actual failure mechanism to the level necessary to correct the problem. When the same failure, contaminant, or other problem has been diagnosed several times, a manufacturer need not analyze all additional gloves that are returned with the same complaint.
When an investigation is made under §820.198(e), a record of the investigation shall be maintained by the manufacturers formally designated unit identified in §820.198(a). The record of investigation shall include:
When the manufacturer's formally designated complaint unit is located at a site separate from the manufacturing establishment, the investigated complaint(s) and the record(s) of investigation shall be reasonably accessible to the manufacturing establishment per §820.198(f). Complaints are required to be accessible to the actual manufacturing site so that quality problems can be identified and corrective action implemented as required by §820.100. If a manufacturer's formally designated complaint unit is located outside of the United States, records required by this section shall be reasonably accessible in the U.S. at either:
Records of non-valid complaints need not be sent to the actual manufacturing site. Relabelers, importers, and others who distribute under their own name should forward complaints to the actual manufacturer. The forwarding of complaints should be considered when developing contracts or other business arrangements with importers.
Per §820.198(d) any complaint that represents an event which must be reported to FDA under 21 CFR parts 803 or 804 shall be promptly reviewed, evaluated, and investigated by a designated individual(s) and shall be maintained in a separate portion of the complaint files or otherwise clearly identified. In addition to the information required by §820.198(e) [the list numbered 1-8 above], records of investigation for MDR events shall include a determination of:
*** SAMPLE PROCEDURE *** Do not use without modifying to meet your needs. In this example, management with executive responsibility is stating what shall be done by their company personnel.
| C O M P A N Y L O G O | Sheet 1 of 5 | |
| Title: Complaint Processing Procedure | SOP Number | |
| Prepared by: | Date Prepared | |
| Approved by: | Date | Rev |
| ECN notes | ||
PURPOSE: To establish and implement a procedure and forms for recording customer complaints, analysis, response, and corrective action.
POLICY: It is the policy of our company that all complaints regarding safety, performance, labeling, or quality of our gloves will be subject to management review and/or investigation and will result in prompt response and corrective action where indicated.
SCOPE / DEFINITION: This policy is applicable to and must be complied with by all personnel who receive a customer complaint, including personnel in Sales and other departments.
We define "complaint" as a written or oral expression of dissatisfaction relative to the identity, labeling, packaging, quality, durability, reliability, safety, biocompatibility, effectiveness, or performance of any glove or other device manufactured by us.
Types of complaints intended to be covered by this policy are as follows:
1. PRODUCT PERFORMANCE: the product in some way does not perform to user’s expectation or to any level of performance conveyed to the customer by printed labeling or verbally by company employees.
2. INTERFACE: the product in some way is difficult or awkward to open or use.
3. PRODUCT SAFETY: all safety complaints are covered by this procedure.
4. PRODUCT APPEARANCE: visual defects inconsistent with the user’s expectations for gloves manufactured by our company.
5. GENERAL COMPLAINTS: order or shipping error delayed or unacceptable response to problems, unfulfilled promises, etc.
FORMS USED: Customer Complaint / Analysis (two-sided) and Complaint Log
PROCEDURE: Upon receipt of a customer complaint, the recipient completes side one of a CUSTOMER COMPLAINT form and if the complaint is written, attaches the complaint letter to the form. The recipient then gives the form, with any attachments, by the next day to the Manager of Quality Assurance.
| *** SAMPLE PROCEDURE Quality Assurance: |
Sheet 2 of 5 |
1. Assigns a sequential complaint number and enters the complaint into the Complaint Log.
2. Determines and notes on the complaint form the person to whom the complaint is to be assigned for investigation and/or corrective action and the date a response is required from the assignee.
3. Notes any specific instructions to the assignee.
4. Distributes a copy to appropriate Department(s) as checked on side 1 of the complaint form.
5. Makes 2 copies of both sides of the in-process form and attachments, and distributes:
Original to the Assignee.
One copy to the "UNDER INVESTIGATION" complaint folder.
The Assignee:
1. Performs the investigation and/or corrective actions and records the results on the form; and attaches any investigation records.
2. Returns the original of the in-process form to QA.
Quality Assurance:
1. Records on the Analysis side:
If no action is taken, the reason for inaction should be recorded on the analysis form.
Any additional corrective action taken or directed by QA.
The nature and date of any response made to the originator or the customer. If this response is written, a copy of the letter or FAX is attached to the analysis form.
The final disposition of the complaint.
QA signature and date.
2. Records the final disposition of the complaint on the complaint log.
3. Files the completed form in the appropriate complaint file for the type of product involved; and discards the copy previously filed in the "UNDER INVESTIGATION" complaint folder.
4. Distributes the complaint log monthly to Staff and specifically involved departments. This log should include a trend of complaints for the month correlated with trends noted in previous months.
| *** SAMPLE RECORD *** |
Sheet 3 of 5 |
| COMPLAINT LOG |
MONTH |
YEAR | |||
| Seq. No. |
Date Rec’d |
Type Glove |
COMPLAINT |
DISPOSITION | |
*** SAMPLE RECORD *** |
|
| CUSTOMER COMPLAINT (Side 1) SEQUENTIAL
COMPLAINT NO. ______________ Glove Type ________________________________________________________________ Catalog Number _____________________________________ Lot Number _____________ Distributor _________________________________________________________________ Complainant _______________________________________________________________ Account Name ______________________________________________________________ Account Address ____________________________________________________________ Complaint Received by _______________________________________________________ Title ______________________________________________ Date Received ___________ |
| By: | [ ] Visit | [ ] Phone | [ ] Letter | [ ] Sales | [ ] Credit Memo | [ ] Other |
| Association With User/Patient
_________________________________________________ COMPLAINT ABOUT: [ ] Pinholes, Tears, Fisheyes, Degradation ________________________________________ [ ] Expiration Date / Shelf Life _________________________________________________ [ ] Pinholes, Tears, Fisheyes, Degradation ________________________________________ [ ] Powder, Lubricant, Tacky __________________________________________________ [ ] Particulates: Type _________________ Location _______________________________ [ ] Packaging ______________________________________________________________ [ ] Sterility ________________________________________________________________ [ ] Labeling _______________________________________________________________ [ ] Thickness, Mold, Appearance, Attributes ______________________________________ [ ] Dermatitis ______________________________________________________________ [ ] Hypersensitivity _________________________________________________________ [ ] Describe Other Defects ____________________________________________________ Comments _________________________________________________________________ _________________________________________________________________________ _________________________________________________________________________ |
| ATTACHMENTS: | [ ] Implicated Sample | [ ] Associated Sample | [ ] Letter |
| Received By QA Mgr. ______________________________ Date
_____________________ Assigned To ______________________________________ Response Due _____________ Instructions ________________________________________________________________ |
| Distribution: | [ ] Quality Control | [ ] Engineering | [ ] Production | [ ] Q |
| *** SAMPLE RECORD *** | Sheet 5 of 5 of Complaint Proced. Number________ |
| COMPLAINT ANALYSIS (side 2) _____________ Sequential
Complaint Number ________ Glove Type ______________________________________ Cat. Number ________________ Date of Complaint Report ___________________________ Lot Number ________________ Name of Complainant _________________________________________________________ Nature of Complaint __________________________________________________________ ASSIGNEE EVALUATION & CONCLUSIONS: ___________________________________ ___________________________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ [ ] Pinholes, Tears, Fisheyes, Degradation _________________________________________ [ ] Powder, Lubricant, Tacky ___________________________________________________ [ ] Particulates ______________________________________________________________ [ ] Labeling/Packaging ________________________________________________________ [ ] Non-Sterile ______________________________________________________________ Thickness, Appearance, Color Attributes of: _______________________________________ [ ] Fingers __________________________________ [ ] Palm ________________________ [ ] Crotch __________________________________ [ ] Cuff/bead _____________________ [ ] Elongation _______________________________ [ ] Tensile ______________________ [ ] Chemical Residues Above Spec. ______________________________________________ [ ] Protein Level Above Spec. __________________________________________________ [ ] Improper Use _____________________________________________________________ [ ] Shipping Damage __________________________________________________________ [ ] Describe Other Defects/Problems _____________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ |
| ACTION: | [ ] Recalled | [ ] Replaced | [ ] Credited | [ ] Sales Follow Up |
| [ ] Letter
___________________________________________________________________ [ ] Referred To ______________________________ for Further Investigation or Correction [ ] NONE. Reason for no action _________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ NOTES: ___________________________________________________________________ ___________________________________________________________________________ FINAL DISPOSITION ________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ Reviewed by: Quality Assurance _______________________________ Date ____________ If requested: Engineering _____________________________________ Date ____________ Production _________________________________________________ Date ____________ |
A quality audit is an independent inspection and review of all aspects of a quality system. This audit covers all of the manufacturers operations that could affect the safety and effectiveness of the device and thus, as appropriate, should also include suppliers, calibration laboratories, and contractors. Audits are described in detail in chapter 17 of the Medical Device Quality Systems Manual: A Small Entity Compliance Guide.
Because the definition of manufacturer includes initial distributors (importers), initial distributors of gloves are subject to the audit requirements for their operations that are covered by the QS regulation. These operations include such activities as environmental control for, and handling and storage of, finished gloves and forwarding complaints to the actual manufacturer.
Gloves are presently made by causing a polymer solution, on a macro and microscopic basis, to completely coat formers. Among several other reasons, this process will consistently produce quality gloves only if the:
Therefore, contamination and environmental control are very important and should receive specific attention during audits. These and a few other specific items to audit are listed below in short trigger sentence/phrase form. These questions should be expanded into multiple questions, as appropriate, and should be asked in addition to typical or basic audit questions.
Audit Checklist. Are there written procedures, and related records for:
CORRECTIVE AND PREVENTIVE ACTION
Analyzing problems, negative and/or exceptional information to solve product or quality system problems is a very important and vital part of a quality system. Without feedback and corrective and preventive action (CAPA) a quality system degenerates into disjointed activities which is not self correcting; and it will, to various extents, be out-of-control. Unfortunately, the manufacturer will not know the extent that the system is out-of-control until a significant problem occurs. (Please see the system diagrams at the beginning of this chapter.)
Corrective and preventive action is required by §820.100. Feedback information for CAPA comes from observations and data derived from routine activities such as design verification, manufacturing data, customer complaints, etc. For example, cut gloves observed during stripping may indicate a protrusion has fallen into the line processing area. Feedback data also comes from directed activities such as audits to search for any deficiencies in the quality system.
To ensure that CAPA is performed, §820.100 lists a series of required activities. Section 820.100 also requires that CAPA activities be documented and that management be informed about quality problems and CAPA. Therefore, management has the information on which to base quality-related decisions.
A first CAPA action should be to make certain that adequate system and product data are being collected and an adequate audit procedure and checklist or other suitable method is being used to assure complete coverage of the audit requirements and the company quality system. Without adequate data, and analysis of the data to find non-conformances, a CAPA program cannot function.
During an inspection, FDA investigators will, on a priority basis, look at management of the quality system and CAPA activities because of their vital importance in maintaining a quality system.
Checklist for the design of gloves and associated processes. It does not cover all possible parameters. It is in keyword form; therefore, it should be used by appropriately trained persons. Do not use this checklist without modifying it to meet your specific approach to glove and process design.
CHECKLIST FOR DESIGN OF NATURAL RUBBER LATEX GLOVES AND PROCESSES

A way to remember the outline of the CAPA requirements is to note that an avid approach to CAPA should be used. This memory association yields:
A Analyze I Investigate I Identify V Verify and validate I Implement D Disseminate
And, of course, quality system activities are documented and management is kept informed
Sample of a letter to pre-announce an FDA International Inspection Letter
Conduct During the Inspection
Close-out Meeting
After the Inspection
Management Letter
Warning Letter
Seizure
Administrative Detention
Restraining Orders and Injunctions
FDA
REQUIREMENTS FOR MEDICAL GLOVES
COMPLIANCE ACTIVITIES
FOR IMPORTED GLOVES
DETENTION
Administrative Detention
Detention Without Physical Examination
Criteria for Detention Without Physical Examination (DWPE) - General
Criteria for Release from Detention Without Physical Examination (DWPE) - General
Import Alerts for Medical Gloves
FDA SAMPLING
EFFORTS
ENFORCEMENT
STRATEGY
Appendix A - Selections From the FDA Regulatory Procedures Manual
Appendix B - Sec.335.700 Surgeons' Gloves and Patient Examination Gloves
Appendix C - 21 CFR, Title 21, Volume 8
FDA determines compliance with the medical device regulations primarily by factory inspections. During the inspection, the FDA investigator will review your facilities, design controls, manufacturing operations, environment, and records to determine your level of compliance. The major emphasis will be on compliance with the Quality System (QS) regulation. Thus, it is important that manufacturers develop, maintain and use a quality system as outlined in this manual.
Manufacturers should be familiar with the QS regulation. Prior to an inspection, you may want to conduct an audit of your manufacturing operations and processes, or have an audit performed by a qualified auditor who is not associated with the firm. Any deficiencies identified should be corrected and implemented prior to the FDA inspection. Manufacturers should not depend on an FDA inspection to do this QS assessment for them. FDA performs pre-announced inspections for firms with no adverse compliance history. After the firm has become familiar with the QS regulation, they should develop an internal plan for dealing with inspections (FDA, ISO, etc.). The plan should detail the manufacturer’s policy regarding inspections, and designate the specific individual(s) who will accompany and/or assist the investigator. Receptionists should be informed when an investigator is scheduled to visit the facility and instructed as to who is to be contacted once the investigator arrives.
Sample of a letter to pre-announce an FDA international inspection. Footnotes added to sample letter.
DEPARTMENT OF HEALTH AND HUMAN
SERVICES
_________________________________________________________________
|
U.S. Food and Drug Administration Telephone: 301-827-5653 or 5632 Number of Pages Sent 2 |
DATE:
TO:
FAX:
ATTN:
Dear _________________
I am writing to pre-announce an inspection of the above referenced firm by an inspector of the U.S. Food and Drug Administration. This will be a ________* inspection covering the FDA's Current Quality System/Good Manufacturing Practice Regulations for Medical Devices (21 CFR Part 820) and is part of our effort to inspect foreign manufacturers that export their products to the United States.
There is no cost to your firm for this inspection. However we do ask that an English speaking member of your staff or interpreter be made available during the inspection.
We are scheduling an inspector to be in your country during the month of _____________. The proposed dates to conduct this inspection are as follows:
Footnotes:
Inspections of medical device firms will cover all applicable sections of 21 CFR Part 820 as they pertain to your firm.
* Usually a GMP inspection for glove
manufacturers.
Design Controls apply to surgeon's gloves per 21 CFR
§820.30(a)(2)(ii).
If this site does not have the research and development or specification developer (i.e., you believe design controls are not applicable), please provide the following information:
R & D or Specification Developer:
Firm Name:
Address:
City, Country:
Contact Person:
Phone
Number:
Fax Number:
Please confirm receipt of this message, and that these dates are suitable by return fax as soon as possible.
Upon confirmation I may also ask for your assistance in arranging for hotel accommodations and transportation to and from your plant on the days of the inspection.
Thank you in advance for your attention to my request and I look forward to hearing from you shortly. If I can be of assistance, please do not hesitate to contact me at 301-827-5632.
Sincerely,
Liliane Brown
Associate Director
*****************************************************************************
Upon arrival at your facility, the investigator will present his/her credentials and issue an FDA 482, Notice of Inspection, to the most responsible person at the facility. FDA does not issue Notices of Inspection for foreign inspections. If a foreign manufacturer refuses an inspection without a valid reason, their devices are adulterated per §820.1(d) and FDA may detain their devices at the U.S. port of entry. The manufacturer should examine the FDA investigator’s credentials. Then, the receptionist or initial contact persons should inform all key employees that an FDA investigator is present.
If the investigator is not familiar with the manufacturer, the contact person should describe the product line and operations, and review their policies and programs with the investigator. During the inspection, the investigator may continuously record his/her observations. The FDA investigator will provide a written list of any significant observations (deviations from the QS Regulation) at the close of the inspection. These observations will be placed on form FDA-483, "Inspectional Observations." If your representative disagrees with any observation made by the FDA investigator, be sure to discuss the reason for the observation with the investigator. You may find that there was a misunderstanding that can easily be corrected. These observations will be discussed with the manufacturer’s management.
During the inspection the FDA investigator may hold a discussion with firm representatives at the end of each day. This provides time for the investigator to tell the firm about any problems or concerns they have with the areas covered during the inspection to date to clarify any misunderstandings that may have occurred.
A company individual should accompany the investigator any time they are in the production areas, reviewing documents, or talking to firm employees about the inspection. Understanding what the investigator is inspecting is an important part of handling an FDA inspection. Comments and suggestions made by the investigator, unanswered questions, and promises should all be recorded. The firm should be taking their own notes on the general areas of the plant visited by the investigator, whom they spoke to, the documents reviewed, and copies. This information can help management to prepare their comments in response to any deviations listed on the form FDA-483 at the conclusion of inspection.
If any records copied by an investigator contain trade secret or otherwise confidential information, these records should be identified, i.e., by a confidential stamp. Do NOT automatically mark every page of a document as confidential. This information is used by FDA in determining if the record may be released under the Freedom of Information Act.
Occasionally during a domestic inspection, the investigator may collect exhibits to document conditions in the factory, or collect samples to verify product quality, or to investigate user complaints. Whenever an investigator collects samples, duplicate samples should be collected and stored by your company. Before leaving your premises with a sample, the investigator will issue a form FDA-484, "Receipt for Samples." Where indicated, the investigator will obtain copies of shipping records to document interstate movement of shipments from which these samples were taken. The investigator will then prepare the appropriate FDA affidavit form (forms FDA 463a, 463, 1664a, or 1664b) which will reference these shipping records. A responsible employee of the firm will be asked to verify, by signature, that the documents referenced in the affidavit pertain to the shipment(s) in question.
Each employee should understand the investigator’s questions before answering. If needed, ask for an explanation. Refer each question to the most suitable employee. Questions should be answered by employees who are knowledgeable in the area related to the question. If there are questions for which you don’t have an immediate answer, make a list of these unanswered questions, get the answers, and give them to the investigator.
During the inspection, the firm can implement corrective action on any QS regulation deficiencies noted by the investigator with which they agree. The investigator should be made aware of any corrections after they are implemented because these corrections will show intent to comply with the FDA regulations.
At the end of a factory inspection, the FDA investigator conducts a close-out meeting. During this meeting, the investigator will discuss the observations recorded on form FDA-483 with the manufacturer's management. Representatives of the firm will be given a copy of the completed form FDA-483, which should be checked for accuracy and completeness against their notes. If no FDA-483 is issued, the investigator will discuss his/her findings in general. Management with executive responsibility should be present to answer questions about any corrective actions to be taken and schedules for these actions. The firm has an opportunity to have the form FDA-483 annotated by the investigator with one of four select annotations based on the firm's responses: corrected and verified; reported corrected, not verified; promised to correct; or no comment. If a firm does not wish to have the form FDA 483 or any particular observation annotated, they should let the investigator know.
The investigator should be reminded of any corrections that have been made. Discuss your plans to make corrections, and provide a tentative schedule for these future actions. Answers given at this meeting will be recorded by the investigator. During the close-out meeting, make sure that all deviations are adequately discussed. If there is disagreement, present all of the manufacturer’s information and any regulations and official FDA interpretations that support your viewpoint.
It is imperative that the manufacturer respond to any recommendations or observations made by the FDA investigator or other official. If you disagree with an observation, include your reason and supporting documentation, regulations, and/or official FDA interpretations. A written response to the form FDA-483, along with documentation to show how the manufacturer has corrected or intends to remove or correct the objectionable conditions or practices, can help assure the FDA that the manufacturer has corrected or intends to correct listed violations. A clear, quick response will demonstrate the manufacturer’s intent to comply with the medical device regulations. You should prepare a response even if you do not receive anything from FDA in writing. To repeat, a plan of corrective action is very important. Domestic manufacturers may also request a meeting with FDA district management to discuss violations and your proposed courses of action. This approach allows you to present your case to the FDA.
Foreign manufacturers should provide their response, including their rationale for any unresolved items to:
FDA/CDRH (HFZ-306)
2094 Gaither Road
Rockville, MD 20850 U.S.A.
A post-inspectional letter will be sent to the firm by FDA indicating the firm’s state of compliance such as no action indicated (NAI) or voluntary action indicated (VAI). FDA will mail a completed Establishment Inspection Report (EIR) to management of the firm after FDA determines that the inspection is closed.
If management with executive responsibility is not present during the issuance of the form FDA-483 at the end of the inspection, FDA may send a "Management Letter" to company management with executive responsibility to assure that they have a copy of the issued form FDA-483. The Management Letter is only a brief transmittal letter, and is not to be confused with the "Warning Letter" described below.
A Warning Letter is a specifically-worded enforcement letter written by top management of an FDA field or headquarters unit to management with executive responsibility for a firm. The letter is sent by FDA to a manufacturer primarily to draw the manufacturer’s attention to violations and thereby, obtain prompt correction. A Warning Letter is intended to obtain correction of deficiencies noted:
A Warning Letter may be issued by FDA instead of immediately seizing the product, obtaining an injunction, or detaining imports. The Warning Letter contains a formal warning to the manufacturer that specific sections of the law have been violated and unless corrective action is taken, the FDA is prepared to impose legal and/or administrative sanctions. Domestic sanctions include seizure, prosecution, injunction, and civil penalties. Unless otherwise indicated, within 15 working days after receiving a Warning Letter, a formal response must be made by the manufacturer to FDA. If you receive a Warning Letter, you should respond by stating the specific steps your organization has taken to correct noted violations, including an explanation of each action to prevent recurrence of similar violations. If corrective action cannot be completed within 15 working days, state the reason for the delay and when the correction will be completed.
A Warning Letter is also considered to be a prior warning and notification to responsible officials of the company of possible civil or criminal action to be taken by FDA.
Responsible individuals should not assume they will always receive a Warning Letter before FDA initiates administrative action or recommends an injunction, seizure, civil penalty and/or criminal proceeding. Before initiating formal regulatory action, FDA is under no legal obligation to warn manufacturers or individuals that they or their products are in violation of the law. For example, the FDA ordinarily will not issue a Warning Letter but will take other action such as seizure and injunction when:
An FDA Warning Letter to a manufacturer does not preclude initiation of other concurrent action, such as seizure or administrative detention, as part of an overall enforcement strategy.
A seizure is a civil court action against a specific quantity of goods whereby FDA seeks to remove these goods from commercial channels. After seizure, no one may move or tamper with the goods except by permission of the court. The owner of the seized merchandise is usually given approximately 30 days by the court to decide on a course of action. If no action is taken and the owner does not file a claim to the goods, the court generally will recommend disposal of the goods. If the owner decides to contest the Government’s charges and files a claim to the goods, the case will be scheduled for trial. A third option allows the owner of the goods to request permission of the court to bring the goods into compliance with the law. In this situation, the owner/claimant of the goods is required to provide a bond (money deposit) to assure that the orders of the court will be performed, and the owner/claimant must pay for FDA supervision of any activities by the manufacturer to bring the goods into compliance.
An administrative detention prohibits the distribution or use of adulterated or misbranded devices encountered during inspections. The detention usually lasts up to 30 days, possibly longer, until FDA has considered what action it should take concerning the devices, or has initiated legal action if appropriate. During the detention, detained devices may not be used, moved, altered, or tampered with in any manner by any person.
Restraining Orders and Injunctions
A "Temporary Restraining Order" (TRO) may be sought by FDA before an injunction and is designed to stop the alleged violative practice until the court can hear evidence that may lead to an injunction. A TRO imposes restraint upon a defendant for not more than 10 days, although this period may be extended by the courts.
An injunction is a court order that restrains a person or manufacturer from violating the law, e.g., to prevent interstate distribution of violative products, and to correct conditions in the establishment in which the violation occurred.
FDA REQUIREMENTS FOR MEDICAL GLOVES
When patient examination gloves and surgeon’s gloves were initially classified by FDA, patient examination gloves were exempted from 510(k) premarket notification and medical device good manufacturing practices.
On January 13, 1989, the FDA published regulations in the Federal Register which removed the exemptions from 510(k) and QS regulatory requirements. On December 12, 1990, regulations were published which specified defect levels for adulteration of patient examination and surgeon’s gloves (21 CFR 800.20). Prior to the passage of this regulation, FDA initiated inspections of glove manufacturers to assure adherence to medical device QS requirements and initiated a comprehensive testing program to assure conformance to acceptable defect levels. FDA sampling methodology and defect action levels are detailed in the FDA Compliance Policy Guides, Chapter 3, Devices, Subchapter 335, General Hospital, Section 335.700, Surgeon’s Gloves and Patient Examination Gloves; Defects - Criteria for Direct Reference Seizure. This guide is included at the end of this chapter.
The FDA test method for gloves is detailed in 21 CFR §800.20, which is reprinted at the end of this chapter. This rule states that FDA’s analysis of gloves for leaks is conducted by a water leak method, using 1000 milliliters (ml) of water. Each medical glove is analyzed independently. When packaged as pairs, each glove is considered separately, and both gloves are analyzed. A defect in one of the gloves is counted as one defect; a defect in both gloves is counted as two defects. Defects are defined as leaks, tears, mold, embedded foreign objects, etc. A leak is defined as the appearance of water on the outside of the glove. This emergence of water from the glove constitutes a watertight barrier failure. Leaks or defects within 1½ inches of the cuff are disregarded. A glove with multiple quality problems such as a hole, mold, embedded foreign objects, etc., is counted as one defective glove.
COMPLIANCE ACTIVITIES FOR IMPORTED GLOVES
The FDA periodically performs inspections of foreign manufacturers, typically with the permission and cooperation of the manufacturer and foreign government. FDA cannot impose the same regulatory sanctions upon foreign manufacturers that it can upon U.S. manufacturers, e.g., injunction or prosecution. If a foreign manufacturer refuses to permit or allow the completion of an FDA inspection, its devices will be considered adulterated under section 50l(h) of the FD&C Act and will be detained at the point of entry [see 21 CFR 820.1(d)].
Section 801 of the FD&C Act details FDA’s authority over imported devices. FDA is authorized to examine samples of incoming medical devices, and refuse entry to products that appear to be adulterated or misbranded. This includes apparent non-compliance with medical device QS requirements as well as sample analyses confirming device defects in excess of specified defect levels. At import, various information will be needed to identify the shipment and to verify its status. To reduce import delays, invoices should reflect the following under "Description of Entry:"
Please note that the FDA Modernization Act of 1997 also requires foreign manufacturers exporting medical devices to the U.S. to be registered (form FDA 2891).
You should also confirm that on invoices, shipping records, etc.:
When a shipment of a foreign manufacturer’s gloves is presented for import, the FDA district office may elect to sample the shipment for testing. When this occurs, the importer of the gloves will receive a Notice of FDA Action. Upon receipt, the importer should contact the detaining district if he wishes to move the shipment to his own premises or to a warehouse of his choice. The importer should not distribute the gloves until they are tested and released. Remember, these gloves are not legally entered into the U. S. until a notice of release is issued by the FDA. If the gloves fail FDA testing, the importer will be asked to account for all gloves in the shipment. If some of the gloves have been distributed and cannot be accounted for, the importer may incur a penalty based upon his/her failure to redeliver the goods to the U.S. Customs Service.
If any lots of medical gloves are found to be adulterated or misbranded, the manufacturer or importer of record must bring them into compliance. He/she should advise the FDA district that initiated the detention of the firm's plans for bringing the product into compliance. If the product cannot be brought into compliance, it cannot be marketed in the U.S. as medical gloves.
Non-conforming medical gloves may be brought into conformance by:
If the manufacturer/importer chooses to recondition the product, they must request and obtain permission from the responsible FDA District Office.
One method of reconditioning out-of-compliance medical gloves may be to label and market them for use in food handling or preparation. Another method may be to label and market such gloves for non-FDA regulated use, such as household gloves, painter’s gloves, etc. If relabeling the product for a non-FDA regulated use, be careful to remove all inferences that the product may be suitable for an FDA regulated use. This may even include modifying reference to an establishment name. For example, Topgrade Medical Glove Supply Corporation or International Hospital Glove Supply Company would not be suitable establishment names on the labeling of gloves being reconditioned for non-FDA regulated use.
Detention is the administrative action taken by FDA in accordance with its regulations at 21 CFR 800.55 against imported medical devices that are not in compliance with the laws which FDA administers. Imported medical devices may be detained whenever physical examination or testing of a medical device, or examination of medical device labeling and labeling claims by FDA reveals the medical device to be in violation of FDA laws. The importer of record may file an appeal requesting an informal hearing at which a presiding FDA officer shall affirm or revoke the detention. Detained devices are either released if brought into compliance, or refused entry if not brought into compliance.
For information about importing and exporting medical devices, please see our International web site at: http://www.fda.gov/cdrh/international/
Detention Without Physical Examination
Detention Without Physical Examination (DWPE) is the administrative act by FDA of detaining the entry of a specified article, usually from a specific supplier, and occasionally from all suppliers from a specific country, without physical examination or testing. DWPE differs from general administrative detention in that it is imposed based on the previous violative history of an imported medical device being offered for entry into the U.S. and does not occur as a result of a violative analysis or elimination of the present entry found by FDA. DWPE is an effective action used against severe or chronic violations or violators. It is also an effective control for those importers that expect the FDA to serve as a quality control laboratory for them. DWPE essentially places the responsibility for determining quality and compliance with the law upon the U.S. importer or broker, and indirectly upon the foreign supplier or sometimes a country. DWPE is generally based on information regarding the past violative history of the medical device and/or other information indicating that the medical device may be violative.
DWPE actions are implemented through the issuance of FDA "Import Alerts." Copies of FDA Import Alerts may be obtained at the FDA webpage under the "Field Programs" link or from the FDA Division of Import Operations and Policy. The medical glove import alert is found at: http://www.fda.gov/ora/fiars/ora_import_ia8004.html
FDA derives its authority to impose DWPE directly from section 801(a) of the Federal Food, Drug, and Cosmetic (FD&C) Act, which states that an article (device) may be refused admission:
If it appears from the examination of such samples or otherwise that:
(1) The methods used in, or the facilities and controls used for, the manufacture, packaging, storage, or installation of the device do not conform to the requirements of 520(f) (Good Manufacturing Practice Requirements)
(2) Such article is forbidden or restricted in sale in the country in which it was exported, or
(3) Such article is adulterated, misbranded, or in violation of Section 505 [New Drugs], then such article shall be refused admission.
It is important to note that the phrase "or otherwise" authorizes refusal of entry or detention on the basis of information other than the results of examination of samples. FDA may consider information such as an article’s violative history as a legally sufficient reason for refusing admission under the FD&C Act. The FDA may use whatever evidence is available to evaluate the potential compliance of the medical devices offered for entry into the U.S.
Criteria for Detention Without Physical Examination (DWPE) - General
Any FDA field office, Center, or other headquarters unit may recommend devices for DWPE or for removal from DWPE when FDA believes that appropriate criteria have been met. The FDA Division of Import Operations and Policy (DIOP) routinely issues medical device specific "Import Alerts" to the FDA field offices which detail criteria for DWPE. These alerts are recommended whenever there is information suggesting that a significant number of shipments of a particular medical device or medical devices offered for import may be violative. The recommendation may be based on such information as the violative history of a medical device, manufacturer, shipper, grower, or geographic area or country. It may also be based upon other information such as knowledge of poor manufacturing or handling practices within a manufacturing facility or geographic area or country. A DWPE may be invoked without any previous detentions if it can be adequately supported by the submitting FDA unit.
In general, FDA recommendations can be based upon one violative sample collected while the medical device is in import status or in domestic channels if:
Criteria for Release from Detention Without Physical Examination (DWPE) - General
In order for the manufacturer/shipper to be removed from the DWPE list, generally FDA must be provided with satisfactory results of sample analyses for the minimum number of consecutive shipments needed to demonstrate compliance with FDA requirements. The documentation should consist of test records (analytical worksheets) from independent qualified U.S. testing laboratories using the FDA analytical worksheet form FDA 431 or equivalent. Testing performed at the manufacturing facility or by private laboratories in the country of origin is unacceptable due to the potential for rapid degradation of gloves during shipment to the U.S. In addition, a manufacturer may provide documented evidence to demonstrate that their manufacturing operation has implemented controls as necessary for continued assurance of medical device quality. The manufacturer may submit the appropriate documentation with a request to be removed from DWPE to the following office in FDA:
Division of Import Operations and Policy (DIOP)
5600 Fishers Lane (HFC-170)
Rockville, Maryland 20857 USA
Phone: 301-443-6553 Fax: 301-594-0413
Importers or subsidiaries may also contact the FDA near the port of entry and speak to someone who is involved with Import Operations. Callers should have their entry numbers available for reference.
In order to demonstrate compliance, manufacturers will be asked to supply a written request to the FDA which documents evidence of the current compliance status of the manufacturer’s devices. Points to consider for inclusion in this written request include:
If FDA agrees that the results of sample analysis or other evidence submitted demonstrate compliance of the manufacturer and/or medical device, the manufacturer, medical device, or country will be removed from Detention Without Physical Examination.
Import Alerts for Medical Gloves
There are currently three major import alerts under which medical gloves are commonly refused entry:
Import Alert #80-04 (Including 3 Levels of Detention for Recidivist Firms and Release Criteria)
A glove manufacturer/shipper will be placed on Import Alert #80-04 as a result of only one violative FDA analysis. The first time this occurs is referred to as Level 1 detention. If a manufacturer is placed on Level 1 detention, all shipments of gloves of the same categories (i.e., patient or surgeon's examination gloves) will be detained without physical examination, i.e., refused entry upon arrival in the U.S. In order to obtain release of the gloves placed on Level 1 or Level 2 detention the owner must provide evidence that the gloves comply with FDA requirements. For example, sample analyses performed by a qualified independent U.S. testing laboratory may be sufficient evidence to obtain admission of a detained shipment while on Level 1 or Level 2. Generally, the results of sample analyses for at least five consecutive shipments entering the U.S. which demonstrate that the gloves are in compliance with FDA requirements may be considered adequate evidence for removal from Level 1 detention.
The second time within a 24-month period that a manufacturer/shipper has a violative FDA or independent laboratory analysis, the firm will be placed on Level 2 detention. In order to be removed from IA #80-04 Level 2 detention, a manufacturer must provide increased evidence of compliance. Generally, the results of sample analyses for at least ten consecutive shipments entering the U.S. which demonstrate that the gloves are in compliance with FDA requirements may be considered adequate evidence for removal from Level 2 detention. In addition, the manufacturer is notified in writing to review their operations for QS requirements prior to shipping further products to the U.S.
The third time within the 24-month period that a manufacturer/shipper has a violative FDA or independent laboratory analysis; the FDA may issue a Warning Letter. If a Warning Letter is issued, the foreign manufacturer will be placed on Level 3 detention. At this level, analytical evidence alone may not be sufficient to show that gloves have been manufactured to meet minimum quality standards. Further evidence, such as an inspection by FDA (or in some instances, when appropriate, inspection performed by a qualified third party), to assess conformance with the QS regulation may be needed in order for a firm to be removed from Level 3 Detention. Shipments of gloves from firms on Level 3 detention may be denied entry until such evidence is provided.
Note: When a firm is on Level 2 detention, only one more violative analysis by FDA or an independent laboratory could result in placing the firm on Level 3 detention. Therefore it is recommended that firms on Level 2 detention perform a comprehensive and objective review of their manufacturing procedures and practices for conformance with the requirements of the QS regulation. If this review shows that corrections to procedures/practices are necessary to ensure gloves of acceptable quality, then such corrections should be made prior to attempting any further entries of gloves into the U.S.
Import Alert #89-04
If a manufacturer is placed on Detention Without Physical Examination for failure to comply with the requirements of the QS regulation, all shipments of medical devices from the specified manufacturing facility will be refused entry upon arrival in the U.S. Similarly, accessories or parts for these medical devices will be detained.
To remove medical devices that were placed on Detention Without Physical Examination due to failure to comply with the QS regulation, manufacturers must:
After FDA has reviewed the documentation explaining the corrective actions that the manufacturer has implemented or intends to implement and has determined that the corrective actions appear to be adequate, FDA will contact the manufacturer by letter. This letter will advise the manufacturer of this determination and that a reinspection will be necessary to verify the implementation of corrective actions. The CDRH Office of Compliance will request the ORO Division of Emergency and Investigations Operations to make the inspection arrangements.
Division of Emergency and Investigations Operations (HFC-130)
5600 Fishers Lane
Rockville, Maryland 20857 USA
Phone: 301-827-5653
FAX: 301-443-6919
After FDA has determined that the manufacturer is in compliance with the QS regulation, the manufacturer will receive a letter from FDA informing them that their medical devices may now be exported to the U.S., and that they have been removed from DWPE list.
Import Alert #89-08
A glove manufacturer may be placed on Import Alert #89-08 for failure to have a 510(k) premarket notification submission on file with the Agency or for failure to have a finding of substantial equivalence at the time of import. Once on Import Alert #89-08, a manufacturer will be unable to import gloves of the type(s) listed on the detention list in the alert (e.g., latex surgeon’s gloves, vinyl examination gloves, powder-free latex examination gloves, etc.) This situation will continue until the Office of Device Evaluation, CDRH issues a substantial equivalence letter covering the gloves in question. The CDRH is responsible for notifying the Division of Import Operations and Policy to remove the firm from Import Alert #89-08.
It is important for foreign manufacturers who export to the U.S. and U.S. importers to understand the concept of FDA sampling, detention, and detention without physical examination. Due to limited agency resources and vast numbers of imported items, it is not possible for FDA to sample and test all imported food, drugs, cosmetics, biologics, and medical devices. Likewise, it may not be possible for an FDA sample to include all portions of, or lots present in, a shipment or container. The FDA’s sampling efforts are not intended to be quality assurance testing for imported medical devices. As a consequence of limited resources, FDA field offices are constantly attempting to apply their resources in a manner to achieve maximum efficiency. One example of improved efficiency is the use of detention without physical examination, especially for repeat violators (recidivist firms).
When a glove shipment includes multiple lots in each container it is considered a commingled shipment and FDA is not obligated to sample individual lots within the shipment. FDA may collect a sample from one or more lots out of commingled lots of gloves from a single container. Typically, FDA samples gloves from only 6 separate cartons. In this sample, FDA will attempt to represent glove sizes as they occur in the overall shipment and will attempt to include several lot numbers if present. However, exact representation in the sample is not required. Remember, all that is required to refuse entry of a shipment is the appearance of adulteration and such an appearance can be derived even from a sample that includes only one lot number or one size. If the sample fails, all lots are suspect and the container will be detained. In order for the importer to obtain release of commingled gloves, the importer must have the container tested lot by lot and identify which lots exceed defect levels. Only those lots which are shown to be in compliance, subject to verification testing by FDA, will be released for distribution in the U. S.
In addition to taking regulatory actions resulting in the refusal to permit entry of violative imported goods, the FDA has developed an enforcement strategy relative to U.S. importers who engage in business practices that appear designed to evade the lawful regulation of imports. This information is detailed in Chapter 9, Import Operations/Actions, of the FDA Regulatory Procedures Manual reprinted as Appendix A that follows.
SELECTIONS FROM THE FDA REGULATORY PROCEDURES
MANUAL
Chapter 9 - Import Operations/Actions
Subchapter - Priority
Enforcement Strategy For Problem Importers
PURPOSE
To provide guidance for dealing with importers or other individuals who engage in business practices that appear designed to evade the lawful regulation of imports. The procedures outlined in this chapter should not be considered all-inclusive, nor are they intended to limit local options. Situations that appear to involve criminal activity (e.g. smuggling, falsification of records) should also be referred to the Office of Criminal Investigations for their information and follow-up, as appropriate.
This guidance represents the agency's current thinking on dealing with problem importers. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public.
Priority attention should be given to firms with a history of any of the following actions:
BACKGROUND
In developing FDA's automated import system, known as the Operational and Administrative System for Import Support (OASIS), the specific forms "May Proceed Notice," "Release Notice," "Notice of Sampling," "Notice of Detention and Hearing," and "Notice of Refusal" have been replaced by the issuing of "Notices of FDA Action," which includes a description of the specific FDA action (May Proceed, Release, Sampling or Intention of Sampling, Detention, or Refusal) identified for the specific line in the entry. The use of the designations "Product May Proceed," "Product Released by FDA," "Product Collected by FDA," "Product Detained by FDA," or "Product Refused Entry by FDA," or similar wording should be considered as meeting the standard, "giving notice thereof to the owner or consignee." (See 21 USC 381(a); 21 CFR 1.94.)
In 1988, the Agency conducted a short-term enforcement operation aimed at determining the disposition of food articles refused admission. Thirteen percent of articles refused admission for non-labeling violations had been distributed in interstate commerce, rather that redelivered for export or destruction.
In 1990, the Agency discovered an importer of ceramic dinnerware circumventing detention without physical examination by declaring the entries as statuary, a non-regulated article.
Between 1990 and 1992, New York District, in conjunction with the U.S. Customs Service, investigated and documented an importer's history of violative practices regarding the importation of frozen seafood products. Practices included repeatedly importing violative articles; falsifying documents and manipulating articles to avoid detention without physical examination; refusing or not permitting timely inspection of entries; importing previously refused articles; and smuggling. As a result of the investigation, in 1992 the firm's president was indicted by the U.S. District Court in New Jersey. He was subsequently convicted on 138 counts for submitting false documents to FDA and for illegally re-importing previously rejected salmonella contaminated seafood. On February 5, 1993, all frozen seafood products imported by the firm were placed on detention without physical examination.
Between 1992 and 1995, Florida District and the Office of Criminal Investigations, in conjunction with the U.S. Customs Service, investigated and documented an importer's history of violative practices regarding the importation and handling of frozen shrimp. Practices included repeatedly importing violative articles; falsifying documents to avoid detention without physical examination; manipulating articles in attempts to have packers removed from detention without physical examination; and laboratory shopping (sending samples of product that is detained without physical examination to different private labs and then submitting to FDA only the analysis which shows the product in compliance, even though the other lab found the product violative). Further, Florida District identified three shipments of shrimp imported by the firm which were seized because of decomposition. Prior to the seizures, the firm attempted to sell the decomposed shrimp, which had been rejected by eight consignees and the National Marine Fisheries Service. The firm also was discovered washing decomposed, imported shrimp with a copper sulfate solution in an attempt to conceal the decomposition. On March 10, 1995, all frozen shrimp imported by the firm was placed on detention without physical examination. As a further result of the investigation, the firm and its top management were indicted by the U.S. District Court in Florida. The firm's vice president was convicted on 12 felony counts, including conspiracy, obstructing justice, violating Customs law, and tainting shrimp and selling it with the intent to defraud and mislead.
APPROACH
The following enforcement approaches have general applicability. They should be considered when dealing with firms engaged in the types of practices listed in the "Purpose" section above, when conventional import coverage and enforcement avenues appear insufficient to address the problem. The approaches include review and approval of reconditioning proposals (FD-766), the use of Warning Letters (sequential, when appropriate), recall, seizure, injunction, or prosecution.
As always, use of enforcement discretion by the district should be considered in determining the appropriate regulatory response. When egregious actions are encountered, a sequential approach may not be appropriate. Also, situations that appear to involve criminal activity (e.g., smuggling, falsification of records) should be referred to the Office of Criminal Investigations for their information and follow-up, as appropriate.
WARNING LETTERS
Issuance of Warning Letters to remind firms of their responsibilities to import articles that comply with the provisions of the Federal Food, Drug, and Cosmetic Act and other laws enforced by FDA, and to assure that only non-violative articles enter domestic commerce in the United States, is often an appropriate first action. (Refer to RPM Subchapter, "Warning Letters.") Warning Letters may be issued to the importer of record, owner, or consignee (if other than the importer of record) with copies to Customs, and may be issued for the following reasons:
The Warning Letter should state that any distribution of refused articles or articles sampled or intended for sampling that were distributed prior to release are in violation of the Federal Food, Drug, and Cosmetic Act or other applicable acts enforced by FDA, and may result in domestic seizure or other sanctions, including injunction or prosecution.
RECONDITIONING PROPOSALS
The Federal Food, Drug, and Cosmetic Act provides that when an article submitted for entry is found to be violative, the importer has the option of exporting it, destroying it, rendering it not subject to the Act, or requesting permission from the agency to attempt to bring it into compliance with the Act.
If the importer of record decides to attempt to recondition a detained article, section 801(b) of the Act (21 USC 381 (a)) provides that the owner or consignee (by practice, FDA also accepts applications from an importer of record, with a properly posted bond, as the agent of the owner or consignee) may submit to the FDA a written application (Form FD-766 or other acceptable means) requesting permission to bring into compliance an article that is adulterated, misbranded, or in violation of Section 505 (see 21 USC 381 (a)(3)). The owner or consignee may bring the article into compliance by relabeling or other action, or by rendering it other than a food, drug, device, or cosmetic. (Refer to RPM Subchapter, "Reconditioning.")
The approval of the reconditioning application is at FDA's discretion. The Agency should require appropriate controls and provisions as a part of any application before it approves the reconditioning. The application is an agreement between the importer (or other appropriate party submitting the application) and the Agency.
If FDA has documented an importer's practice of consistently importing violative articles not already subject to detention without physical examination and only attempting to recondition the articles after detention, the District may require, as part of any reconditioning application, that the importer agree to destroy any article not brought into compliance during reconditioning, in lieu of permitting re-export of the violative article.
Districts should consult and obtain the concurrence of both the ORO/Division of Import Operations and Policy (DIOP) and the appropriate Center Compliance Office before initiating a policy requiring a specific importer to destroy rather than re-export violative articles as part of every reconditioning process. The information supplied should include, but not be limited to, the following:
REQUESTS FOR VOLUNTARY RECALLS
Although requests for voluntary recalls duplicate a request for redelivery action to some degree, they also offer definite advantages. Experience indicates that requesting the firm to initiate a voluntary action, such as a recall, may result in a more favorable response by the firm than a demand for redelivery. A recall may occur more promptly because it can be initiated in a matter of days, while redelivery may not take place for 90 days or more. This is especially significant in hazard-to-health situations. A recall may provide FDA with further knowledge of the status of the violative merchandise being returned and usually makes it easier to maintain control of the article. This ultimately leads to improved consumer protection.
District management should very carefully encourage the firm to consider a voluntary recall under the following situations:
When an importer fails to respond fully or in a timely manner to a Warning Letter, or we are notified by Customs that an Importer has not responded to a Notice of FDA Action Specifying Refusal of the product, it may be an indication the goods are no longer intact. A visit to the importer may be appropriate and, if articles are missing, attempt to determine the firm's intentions with respect to corrective action.
When a potential health hazard situation exists and the article has been illegally distributed, appropriate press coverage may issue naming firm, product, and country of origin. Issuance of all publicity must be in accordance with guidelines.
Import recalls are to be conducted in full accordance with the guidelines in RPM Subchapter, "Recall Procedures." Supervision of the disposition of returned articles may be made either by FDA or Customs. If disposition will be by destruction, it is suggested that FDA provide the supervision. If the articles are to be exported, Customs or FDA may handle the supervision.
SEIZURE
Seizure is another enforcement approach that may be considered to gain control over violative imported articles. Seizure is an action against an article. Consequently, it will be necessary to show, through laboratory analysis or otherwise, that the article seized is actually violative. An importer's history of illegal actions, while relevant, is not itself sufficient to support seizure. Whatever the importer's previous history, it will be necessary to show that the article itself is violative.
Seizure may be considered for an article which:
When an imported article is seized, and condemned, it is subject to the provisions of section 304(d) (21 USC 334(d)) which may allow for re-exportation of the article, provided specified conditions are met. Under 21 USC 334(d), certain condemned imported articles may be re-exported under limited circumstances. Re-exportation is not available for condemned unapproved new drugs (see 21 USC 355), or foods in violation of the emergency permit control provision (see 21 USC 344). Such articles must be destroyed.
In order to be able to re-export condemned imported articles, the party seeking re-export must satisfy several threshold conditions:
The violation did not occur after the article was imported.
The party seeking re-export "had no cause for believing that it was adulterated, misbranded, or in violation before it was released from Customs custody."
The party seeking re-export must "establish that the article was intended for export at the timethe article entered commerce." An example of where it may be possible to demonstrate that a product was intended for export at the time it entered commerce would be when products are imported for purpose of transshipment to a destination outside the U.S.
a. Intended for export.
b. Accords with the specifications of the foreign purchaser (unless the article is to be exported to the original foreign supplier, in which case there is no need to comply with this requirement).
c. May not be in conflict with the laws of the country to which it is intended for export unless the article is to be exported to the original foreign supplier, in which case there is no need to comply with this requirement).
d. Labeled on the outside of the shipping package that it is intended for export.
e. Not sold or offered for sale in domestic commerce.
Therefore, there are circumstances where the seizure of an article may not accomplish more than detention and refusal of the article, other than stricter control over the goods before re-export and compliance with the applicable requirements of Section 801(e) (21 USC 381(e).
Consequently, in evaluating whether a seizure is an appropriate course of action, a district should consider whether the facts in the case would justify recommending to a court that re-export of the article would be an unsatisfactory resolution. Among the points to consider are:
Under certain circumstances, the district may recommend seizure of violative articles under 21 USC 334 while the articles are still under import status, rather than allow re-export as provided under 21 USC 381 (a). Generally, seizure of articles while in import status may be appropriate if the articles must be destroyed (pose a serious health hazard or it is likely that the articles will be reintroduced into the United States), or the public health requires that certain conditions be imposed (e.g., conditions in 21 USC 381(e)(1)).
As with citation, prosecution, and injunction, samples collected for seizure consideration should, whenever possible, include a 702(b) portion (see 21 USC 372 (b)). Such samples should be collected, sealed, analyzed, and otherwise handled in accordance with procedures normally applied to domestic samples.
State embargo authority and Customs holds are alternative methods to gain control over violative articles. Customs may also release an article at our request so that an immediate domestic seizure may be conducted. Moreover, if a violative article represents evidence of a crime, it may be seized pursuant to a criminal search and seizure warrant. These avenues should also be considered, especially if an importer is likely to attempt to quickly re-export the article.
INJUNCTION
If injunction is the action of choice, the case should be developed in accordance with the procedures set forth in RPM Subchapter, "Injunctions." Injunctions may require a pattern of actual violations with some recognizable danger of a recurrence. The monitoring of an injunction is resource intensive. These facts should be taken into consideration when evaluating this course of action. Also consider that an injunction often results in a hearing more quickly than does a prosecution, particularly if a Temporary Restraining Order (TRO) is requested. This can result in quick corrective action as well as more rapid and efficient redelivery if this response is requested in the injunction. Also, the burden of proof is less in civil cases than in criminal cases, and injunction does not preclude subsequent prosecution for the same violation.
When developing an injunction case against an importer or consignee, there must be a well-documented history of an illegal practice.
A TRO requires a heightened showing of harm. See RPM Subchapter, "Injunctions" regarding the prerequisites for a TRO in conjunction with an injunction action.
CITATION/PROSECUTION
Citation/prosecution should be used when conventional import enforcement approaches are determined to be inadequate to correct violative practices, or the violation is sufficiently egregious to warrant punishment.
When citation/prosecution is the action of choice, refer to RPM Subchapters, "Citations" and "Prosecution" for the appropriate procedures.
Districts should consider the potential impact of developing citation/prosecution recommendations as the action of choice in the following instances:
This list is not all inclusive and there may be other situations where citation/prosecution is appropriate.
Any recommendation for citation, prosecution, or injunction must be supported by fully documented instances of attempts to circumvent normal import procedures. For a felony prosecution recommendation, there must be a fully documented attempt to do the same, with evidence of the intent to defraud or mislead. It is not necessary, in developing a citation/prosecution recommendation, to show that each specific entry is actually violative. However, physical evidence that documents the violative nature of an entry (or of several entries) would be useful to highlight the likely result of the firm's pattern of behavior.
It is important to remember that sample collection and analytical procedures in these cases, as for seizures and injunctions, should differ from routine import work. The Office of Chief Counsel has consistently advised us that when an import physical sample is collected for use in an anticipated legal action, a sealed 702(b) portion should be available (21 USC 372 (b). This request is further supported by guidance provided in the RPM. Proper chain of custody should also be maintained for these samples. Ordinarily, check analyses should be conducted on such samples. In instances where Compliance Policy Guides exist and instructions differ for domestic legal actions as opposed to import detention, districts should follow the guidance for domestic legal actions in terms of types of analyses, check analyses, etc.
Importers of articles detained without physical examination should not feel free to distribute and sell such articles without risk of criminal penalty. Criminal action may be possible against importers violating FDA's detention without physical examination actions or who routinely ship articles without a Notice of FDA Action indicating the articles are Released. Refusal to allow inspection is a violation of the Federal Food, Drug, and Cosmetic Act. Subsequent entry pursuant to an inspection warrant may yield evidence providing the basis for a felony violation for refusal to allow inspection. Distribution of an article prior to receipt of a Notice of FDA Action indicating the article May Proceed or is Released should be considered refusal to permit inspection, as authorized by section 704 (21 USC 374).
In addition to charges under the Federal Food, Drug, and Cosmetic Act and Customs law, Title 19 (note especially, 19 USC 1592 and 1595a), and/or Title 18 charges may also be considered. These include 18 USC 1001, false statements; 18 USC 1505, obstruction of justice (when a firm knowingly and willingly interferes with an FDA inspection by distributing imported articles not released by FDA from import status); 18 USC 542, entry by use of a false statement; 18 USC 545, smuggling; and 18 USC 371, conspiracy.
The following is an excerpt from FDA Compliance Policy Guides, Chapter 3 - Devices.
SEC. 335.700 SURGEONS' GLOVES AND PATIENT EXAMINATION
GLOVES;
DEFECTS - CRITERIA FOR DIRECT REFERENCE SEIZURE (CPG
7124.31)
BACKGROUND:
Surgeon's and patient examination gloves have been increasingly relied upon by health care workers as a barrier to the transmission of Human Immunodeficiency Virus (HIV) and other blood and fluid-borne infectious agents. On August 21, 1987, the Centers for Disease Control recommended that health care workers wear medical gloves routinely because of the potential for transmission of HIV between patients and health care workers. Because hard to detect glove defects, such as holes, can compromise the effectiveness of the glove barrier and pose risk to the health of both patients and health care workers, FDA issued guidelines to the field districts on September 28, 1988, to sample and analyze surgeon's and patient examination gloves of both domestic and foreign origin. Gloves were leak tested using the 1000 ml water method. Regulatory actions under existing authority, such as seizures and detentions of specific glove lots, were handled on a case by case basis. Surgeon's glove lots with failure rates of 10% (10 units in 100) or higher, and patient examination gloves with failure rates of 20% (20 units in 100) or higher were subject to regulatory action. In view of the rapid increase in demand for imported and domestically produced gloves, and the public health benefits of further reducing the risk of transmission of HIV and other blood and fluid borne infectious agents, and to better utilize Agency resources, on November 21, 1989, FDA published in the Federal Register proposed rules to insure that manufacturers of gloves manufacture gloves that are not adulterated. The final rule was published on December 12, 1990, at 55 FR 51254.
FDA will collect samples from lots of gloves to perform the test for defects by the water leak method using 1000 mL water as described in paragraph (b) Test Method of the final rule entitled "Patient examination gloves and surgeon's gloves; sample plans and test method for leakage defects; adulteration." 55 FR 51256 - 51258; 21 CFR 800.20.
The sampling inspection plan used by the FDA has been derived from MIL-STD-105E (the military standard for "Sampling Procedures and Tables for Inspection by Attributes"), based on general inspection level II, normal inspection, and an acceptable quality level (AQL) of 2.5% for surgeon's gloves and 4.0% for patient examination gloves. Single sampling will be used for lots less than or equal to 1200 gloves, while multiple sampling will be used for larger lots. [The FDA sampling inspection plan is described below in 21 CFR 800.20 (c).]
POLICY:
Surgeon's gloves and patient examination gloves that contain holes are adulterated devices. Adulteration will be determined on a lot by lot basis for enforcement purposes. [See 21 CFR 800.20.] Surgeon's gloves whose leakage defect rate exceeds an AQL of 2.5% and patient examination gloves whose leakage defect rate exceeds an AQL of 4.0% will be deemed actionable as described in 21 CFR 800.
REGULATORY ACTION GUIDANCE:
Lots of surgeon's and patient examination gloves that fail the criteria in Attachment A "Sampling Inspection Plan" are subject to direct reference seizure. Districts should forward seizure recommendations to the Division of Compliance Management and Operations (HFC-210).
SPECIMEN CHARGES:
NOTE: Complaints for the seizure of devices should not include allegations of shipment in interstate commerce because allegations of interstate commerce are not required to support seizure of devices [see section 304(a)(2)] .
For lots of surgeon's gloves which are found to be defective at an AQL greater than 2.5%, and for lots of patient examination gloves which are found to be defective at an AQL greater than 4.0% charge:
"The article is deemed adulterated within the meaning of the Act, 21 U.S.C. 351(c) because the quality of the gloves falls below that which it purports or is represented to possess in that the defect rate of the gloves exceeds the permissible rate identified at 21 CFR 800.20."
The proposed letter to the U.S. Attorney should also include the following two paragraphs (fill in the blanks with the appropriate numbers):
Examination gloves are intended for use by health professionals such as physicians and dentists during routine medical and dental examinations. Health professionals rely on examination gloves to prevent the transmission and spread of disease. This has become increasingly important in light of the current AIDS epidemic.
We request seizure because analysis of the gloves by the Food and Drug Administration (FDA) shows that their quality falls below that which it purports and is represented to possess because the defect rate of the gloves exceeds the permissible level as set forth in 21 CFR. ___ out of ___ gloves tested were found to leak or contain holes. 21 U.S.C. 351(c).
The following is taken from 21 CFR Part 800 and contains sample plans and test methods for leakage defects and adulteration of patient examination gloves and surgeon's gloves.
Appendix C
21 CFR, TITLE 21, VOLUME 8
1
[Code
of Federal Regulations]
[Title 21, Volume 8, Food and Drugs, Parts 800 to
1299]
[Revised as of April 1, 1996]
From the U.S. Government Printing
Office via GPO Access
[CITE: 21 CFR 800.20]
TITLE 21--FOOD AND DRUG ADMINISTRATION, DEPARTMENT OF HEALTH AND HUMAN SERVICES--(Continued)
PART 800 --GENERAL
Subpart B--Requirements for Specific Medical Devices
Sec. 800.20 Patient examination gloves and surgeons' gloves; sample plans and \ test method for leakage defects; adulteration.
(a) Purpose. The prevalence of human immunodeficiency virus (HIV), which causes acquired immune deficiency syndrome (AIDS), and its risk of transmission in the health care context, have caused the Food and Drug Administration (FDA) to look more closely at the quality control of barrier devices, such as surgeons' gloves and patient examination gloves (collectively known as medical gloves) to reduce the risk of transmission of HIV and other blood-borne infectious diseases. The Centers for Disease Control (CDC) recommend that health care workers wear medical gloves to reduce the risk of transmission of HIV and other blood-borne infectious diseases. The CDC recommends that health care workers wear medical gloves when touching blood or other body fluids, mucous membranes, or nonintact skin of all patients; when handling items or surfaces soiled with blood or other body fluids; and when performing venipuncture and other vascular access procedures. Among other things, CDC's recommendation that health care providers wear medical gloves demonstrates the proposition that devices labeled as medical gloves purport to be and are represented to be effective barriers against the transmission of blood- and fluid-borne pathogens. Therefore, FDA, through this regulation, is defining adulteration for patient examination and surgeons' gloves as a means of assuring safe and effective devices.
_____________________
1Sketches of
the test apparatus described in this Federal Register are shown in Fig 1 and Fig
2 at the end of the FR text.
(1) For a description of a patient examination glove, see Sec. 880.6250. Finger cots, however, are excluded from the test method and sample plans in paragraphs (b) and (c) of this section.
(2) For a description of a surgeons' glove, see Sec. 878.4460 of this chapter.
(b) Test method. For the purposes of this regulation, FDA's analysis of gloves for leaks will be conducted by a water leak method, using 1,000 milliliters (mL) of water. Each medical glove will be analyzed independently. When packaged as pairs, each glove is considered separately, and both gloves will be analyzed. A defect on one of the gloves is counted as one defect; a defect in both gloves is counted as two defects. Defects are defined as leaks, tears, mold, embedded foreign objects, etc. A leak is defined as the appearance of water on the outside of the glove. This emergence of water from the glove constitutes a watertight barrier failure. Leaks within 1 and \1/2\ inches of the cuff are to be disregarded.
(1) The following materials are required for testing: A 2\3/8\-inch by 15-inch (clear) plastic cylinder with a hook on one end and a mark scored 1\1/2\ inches from the other end (a cylinder of another size may be used if it accommodates both cuff diameter and any water above the glove capacity); elastic strapping with Velcro or other fastening material; automatic water-dispensing apparatus or manual device capable of delivering 1,000 mL of water; a stand with horizontal rod for hanging the hook end of the plastic tube. The support rod must be capable of holding the weight of the total number of gloves that will be suspended at any one time, e.g., five gloves suspended will weigh about 11 pounds.
(2) The following methodology is used: Examine the sample and identify code/ lot number, size, and brand as appropriate. Examine gloves for defects as follows: carefully remove the glove from the wrapper, box, etc., visually examining each glove for defects. Visual defects in the top 1\1/2\ inches of a glove will not be counted as a defect for the purposes of this rule. Visually defective gloves do not require further testing but are to be included in the total number of defective gloves counted for the sample. Attach the glove to the plastic fill tube by bringing the cuff end to the 1\1/2\-inch mark and fastening with elastic strapping to make a watertight seal. Add 1,000 mL of room temperature water (i.e., 20 deg. C to 30 deg. C) into the open end of the fill tube. The water shall pass freely into the glove. (With some larger sizes of long-cuffed surgeons' gloves, the water level may reach only the base of the thumb. With some smaller gloves, the water level may extend several inches up the fill tube.)
(3) Immediately after adding the water, examine the glove for water leaks. Do not squeeze the glove; use only minimal manipulation to spread the fingers to check for leaks. Water drops may be blotted to confirm leaking. If the glove does not leak immediately, keep the glove/filling tube assembly upright and hang the assembly vertically from the horizontal rod, using the wire hook on the open end of the fill tube (do not support the filled glove while transferring). Make a second observation for leaks 2 minutes after addition of the water to the glove. Use only minimal manipulation of the fingers to check for leaks. Record the number of defective gloves.
(c) Sample plans. FDA will collect samples from lots of gloves to perform the test for defects described in paragraph (b) of this section in accordance with FDA's sampling inspection plans which are based on the tables of MIL-STD-105E (the military sampling standard, ``Sampling Procedures and Tables for Inspection by Attributes,'' May 10, 1989). Based on the acceptable quality levels found in this standard, FDA has defined adulteration as follows: 2.5 or higher for surgeons' gloves and 4.0 or higher for patient examination gloves at a general inspection level II. FDA will use single normal sampling for lots of 1,200 gloves or less and multiple normal sampling for all larger lots. For convenience, the sample plans (sample size and accept/reject numbers) are shown in the following tables:
Adulteration Level at 2.5 for Surgeons' Gloves
|
| |||||
| Number defective | |||||
| Lot size | Sample |
Sample |
Number examined |
Accept | Reject |
|
| |||||
| 35,001 and above | First | 125 | 125 | 2 | 9 |
| Second | 125 | 250 | 7 | 14 | |
| Third | 125 | 375 | 13 | 19 | |
| Fourth | 125 | 500 | 19 | 25 | |
| Fifth | 125 | 625 | 25 | 29 | |
| Sixth | 125 | 750 | 31 | 33 | |
| Seventh | 125 | 875 | 37 | 38 | |
35,000 to 10,001 |
First |
80 |
80 |
1 |
7 |
| Second | 80 | 160 | 4 | 10 | |
| Third | 80 | 240 | 8 | 13 | |
| Fourth | 80 | 320 | 12 | 17 | |
| Fifth | 80 | 400 | 17 | 20 | |
| Sixth | 80 | 480 | 21 | 23 | |
| Seventh | 80 | 560 | 25 | 26 | |
10,000 to 3,201 |
First |
50 |
50 |
0 |
5 |
| Second | 50 | 100 | 3 | 8 | |
| Third | 50 | 150 | 6 | 10 | |
| Fourth | 50 | 200 | 8 | 13 | |
| Fifth | 50 | 250 | 11 | 15 | |
| Sixth | 50 | 300 | 14 | 17 | |
| Seventh | 50 | 350 | 18 | 19 | |
3,200 to 1,201 |
First |
32 |
32 |
0 |
4 |
| Second | 32 | 64 | 1 | 6 | |
| Third | 32 | 96 | 3 | 8 | |
| Fourth | 32 | 128 | 5 | 10 | |
| Fifth | 32 | 160 | 7 | 11 | |
| Sixth | 32 | 192 | 10 | 12 | |
| Seventh | 32 | 224 | 13 | 14 | |
1,200 to 501 |
ingle sample |
80 |
5 |
6 | |
| 500 to 281 | Single sample | 50 | 3 | 4 | |
| 280 to 151 | Single sample | 32 | 2 | 3 | |
| 150 to 51 | Single sample | 20 | 1 | 2 | |
| 50 to 0 | Single sample | 5 | 0 | 1 | |
Adulteration Level at 4.0 for Patient Examination Gloves
|
| |||||
| Number defective | |||||
| Lot size | Sample |
Sample |
Number examined |
Accept | Reject |
|
| |||||
| 10,001 and above | First | 80 | 80 | 2 | 9 |
| Second | 80 | 160 | 7 | 14 | |
| Third | 80 | 240 | 13 | 19 | |
| Fourth | 80 | 320 | 19 | 25 | |
| Fifth | 80 | 400 | 25 | 29 | |
| Sixth | 80 | 480 | 31 | 33 | |
| Seventh | 80 | 560 | 37 | 38 | |
10,000 to 3,201 |
First |
50 |
50 |
1 |
7 |
| Second | 50 | 100 | 4 | 10 | |
| Third | 50 | 150 | 8 | 13 | |
| Fourth | 50 | 200 | 12 | 17 | |
| Fifth | 50 | 250 | 17 | 20 | |
| Sixth | 50 | 300 | 21 | 23 | |
| Seventh | 50 | 350 | 25 | 26 | |
3,200 to 1,201 |
First |
32 |
32 |
0 |
5 |
| Second | 32 | 64 | 3 | 8 | |
| Third | 32 | 96 | 6 | 10 | |
| Fourth | 32 | 128 | 8 | 13 | |
| Fifth | 32 | 160 | 11 | 15 | |
| Sixth | 32 | 192 | 14 | 17 | |
| Seventh | 32 | 224 | 18 | 19 | |
1,200 to 501 |
Single sample |
80 |
7 |
8 | |
| Single sample | 50 | 5 | 6 | ||
| Single sample | 32 | 3 | 4 | ||
| Single sample | 20 | 2 | 3 | ||
| Single sample | 13 | 1 | 2 | ||
| Single sample | 3 | 0 | 1 | ||
|
| |||||
(d) Lots of gloves which are tested and rejected using the test method according to paragraph (b) of this section, are adulterated within the meaning of section 501(c) of the Federal Food, Drug, and Cosmetic Act, and are subject to regulatory action, such as detention of imported products and seizure of domestic products. [55 FR 51256, Dec. 12, 1990]
Figure 1
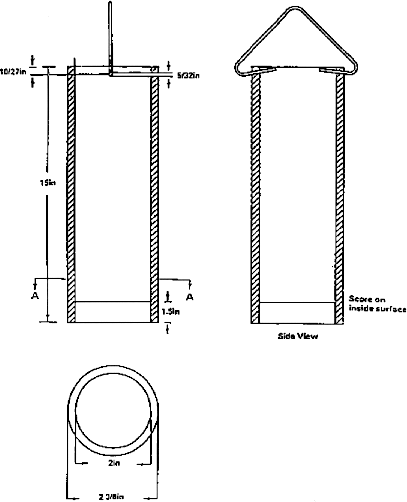
Figure 2
INTRODUCTION TO
STANDARDS
USE OF GLOVE RELATED
STANDARDS
Latex Patient Examination Glove Standards
Synthetic Material Patient Examination Glove Standards
Latex Surgical Glove Standards
Many domestic and international consensus standards address aspects of safety and/or effectiveness relevant to medical devices. Many of these standards were developed with the participation of CDRH staff.
For more information on the use of standards by CDRH, please see http://www.fda.gov/cdrh/stdsprog.html or phone CDRH Facts On Demand at 1-800-899-0381 or 301-827-0111 and specify #321 when prompted for the document shelf number.
CDRH believes that conformance with consensus standards can provide a reasonable assurance of safety and/or effectiveness for many applicable aspects of medical devices. Therefore, information submitted on conformance with such standards will have a direct bearing on safety and effectiveness determinations. In the case of 510(k) submissions for gloves, information on conformance with consensus standards will help establish the equivalence of a new glove to a legally marketed predicate glove for the parameters or areas covered by the standards the manufacturer is meeting.
USE OF GLOVE RELATED STANDARDS
FDA relies on the voluntary standards issued by the American Society for Testing and Materials (ASTM) D 3578, D 3772 (finger cots), D 5250 for the parameters of patient examination gloves and D 3577 for surgeon's gloves. The ASTM website is: http://www.astm.org/. ASTM D 5712 covers the Standard Test Method for the Analysis of Protein in Natural Rubber and Its Products.
ASTM D 6124 covers the Standard Test Method for Residual Powder on Medical Gloves.
ASTM standards are available from:
American Society for Testing and Materials
100 Barr Harbor Drive
West Conshohocken PA 19428 USA
Phone: 610-832-9500 FAX: 610-832-9555ASTM standards are also available from:
Singapore Productivity Board
1, Science Park Drive
Singapore 118221
Phone: 65-278-6666 FAX: 65-278-6665 Website: http://www.psb.gov.sg/
The ASTM standard for each type of glove is noted in appropriate sections of this manual.
Each manufacturer that distributes patient examination or surgeon's gloves in the U.S. should have an original copy of each ASTM or equivalent standard on file referred to by the manufacturer's QS device master record(s) and/or 510(k) submission(s). During an inspection, the FDA investigator may ask to see a copy of each referenced standard.
Manufacturers that want to perform tests for particulates, extractable materials, chemical resistance, bioburden, etc., may refer to IES-RP-CC-005-87-T for Cleanroom Gloves and Finger Cots. This standard is available from:
Institute of Environmental Sciences
940 East Northwest Highway
Mount Prospect, Illinois 60056 USA
Phone 708-255-1561
The following pages present tables of selected data from glove standards around the world. The information presented here is not complete and interested readers should refer to a current copy of the standard for official parameters and other pertinent information.
This information is provided for your reference. The United States FDA may not recognize these standards in whole or in part. Information regarding the glove standards which the FDA does recognize is found elsewhere in this chapter.
The presence or absence of a standard in these tables does not indicate FDA recognition or disapproval of any particular standard.
Latex Patient Examination Glove Standards
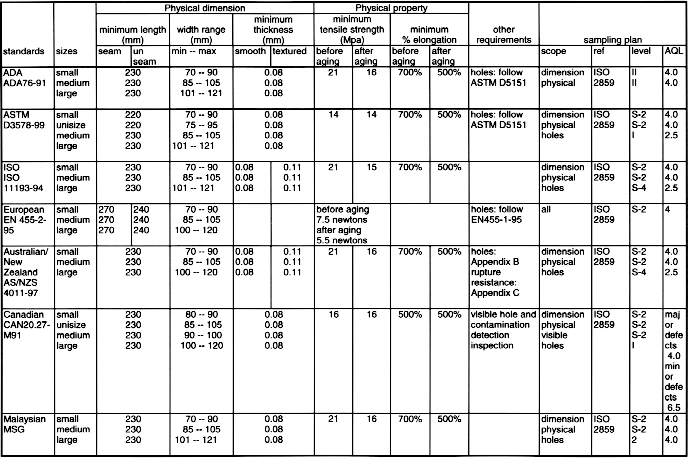
|
latex exam glove standards //ltx exm glv std |
Synthetic Material Patient Examination Glove Standards
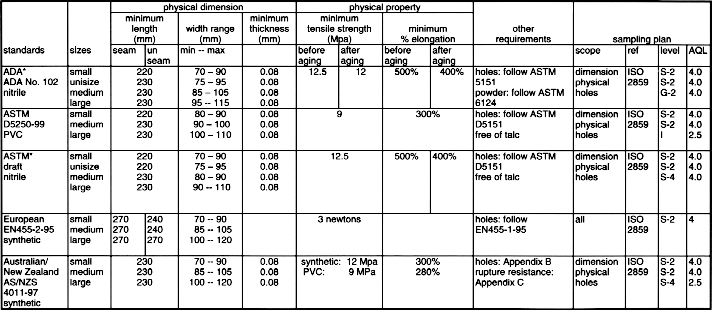
| * : draft standard |
Synthetic Examination Glove Standards syn exm glv std |
Latex Surgical Glove Standards
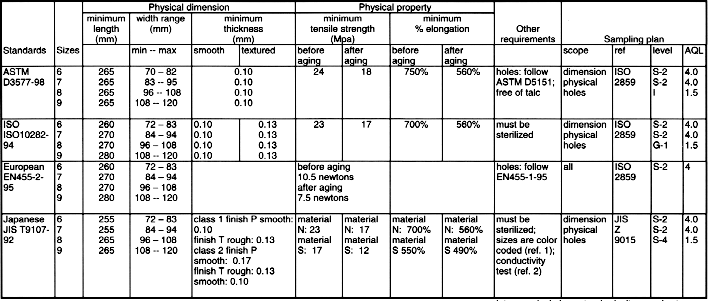
|
latex surgical glove standards ltx srg glv st |
| ANSI/ADA76-91 | Non-sterile latex gloves for dentistry. |
| ADA Spec No. 102 | Non-sterile nitrile gloves for dentistry. |
| ASTM D3577-91 | Standard specification for rubber surgical gloves. |
| ASTM D3578-95 | Standard specification for rubber examination gloves. |
| ASTM D5250-92 | Standard specification for poly(vinyl chloride) gloves for medical application. |
| ASTM draft | Standard specification for nitrile examination gloves for medical application. |
| ISO10282-94 | Single-use surgical rubber gloves-Specification. |
| ISO11193-94 | Single-use rubber examination gloves-Specification. |
| AS/NZS 4011:1997 | Single-use examination gloves—specification. |
| EN 455-1:1995 | Medical gloves for single use. Part 1. Specification for freedom from holes. |
| EN 455-2:1995 | Medical gloves for single use. Part 2. Specification for physical properties. |
| CAN 20.27-M91 | Sterile or non-sterile medical examination gloves for single use. |
| MS1155-89 | Malaysian standard for rubber examination glove. |
| JIS T 9107-92 | Japanese Industrial Standard. Surgical gloves. |
ADA: |
American Dental Association |
| ASTM: | American Society for Testing and Materials |
| CAN: | National Standard of Canada |
| EN: | European Standard |
| ISO: | International Standard |
Other sizes are also available in many standards. Only common sizes are
considered here.
Length is the over-all length and is the minimum
requirement.
Width is the palm width and is always required with tolerances.
Physical requirements are expressed in tensile strength in megapascals and
in ultimate % elongation at break. These are minimum requirements. European
standards require minimum force at break expressed in newtons.
| ASTM D412 | Test methods for vulcanized rubber and thermoplastic rubbers and thermoplastic elastomers—tension. |
| ASTM D573 | Test method for rubber—deterioration in an air oven. |
| ASTM D3767 | Practice for Rubber—measurement of dimensions. |
| ASTM D5151 | Test method for detection of holes in medical gloves. |
| ISO 2859 | Sampling procedures and tables for inspection by attributes. |
| ISO 37-94 | Method for determination of tensile stress-strain properties. (to determine the force at break) |
| ISO 188 | Heat resistance and accelerated aging tests. |
| ISO 4648 | Physical testing of rubber. Methods for the determination of dimensions of test pieces and products for test purpose. |
Applicable glove standards legends //stds legends
APPENDIX
WATER LEAK (PINHOLE)
TESTING LABORATORIES
The following list was compiled as an aid to the medical device industry. An attempt was made to compile an inclusive list from available public information sources. Inclusion of the name of a manufacturer on this list does not connote FDA recognition of their ability to adequately perform the services listed. As with any supplier of raw materials or services, it is the manufacturer’s responsibility to determine and verify the adequacy of the services offered. See Chapter 10 and 21 CFR 820.30 and 820.50.
| Akron Development Labs 300 Kenmore Blvd Akron, OH 44301 |
Ph: (330) 794-6600 FAX: (330) 794-6610 E-Mail: harryb@ardl.com |
William C. Lowenkamp Lowenkamp International P.O. Box 878 Hazelhurst, MS 39083 |
Ph: (601) 894-2802 (601) 894-5873 FAX: (601) 894-2802 E-Mail: Lionfight@Juno.com |
ANRESCO and Microtracers Headquarters 1370 Van Dyke Avenue San Francisco, CA 94124 |
Ph: (415) 822-1100 FAX: (415) 822-6615 Toll Free: (800) 359-0920 E-Mail: Anresco@aol.com |
ANRESCO 790 Basin Street, #2 San Pedro, CA 90731 |
Ph: (888) 359-0920 FAX: (310) 833-4598 |
ANRESCO 2310 NW 55th Court, #123 and 124 Fort Lauderdale, FL 33309 |
Ph: (954) 486-2233 FAX: (954) 486-2886 |
North American Science Associations, Inc. (NAmSA) 9 Morgan Irvine, CA 92618 (only site does water testing) |
Ph: (949) 951-3110 FAX: (949) 951-3280 E-Mail: Jhaase@NAmSA.com |
Updated 4/12/2004
![]()
CDRH Home Page | CDRH
A-Z Index | Contact CDRH
| Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH
